Tratamiento de gliomas (incluso astrocitomas) y de tumores neuronales o glioneuronales infantiles (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Información general sobre los gliomas, los astrocitomas y los tumores neuronales o glioneuronales infantiles
Los tumores encefálicos primarios, que incluyen los gliomas, son un grupo diverso de enfermedades que en conjunto constituyen los tumores sólidos más frecuentes en la niñez. Los tumores de encéfalo (también llamados tumores encefálicos, tumores cerebrales o cánceres cerebrales) se clasifican según sus características histológicas y moleculares, pero la localización del tumor y la extensión de la diseminación también son factores importantes que afectan el tratamiento y el pronóstico. Las características histológicas, el análisis inmunohistoquímico y los hallazgos citogenéticos y genético-moleculares se utilizan en el diagnóstico y la clasificación tumoral.
Se piensa que los gliomas surgen de las células madre y progenitoras neurales que se encuentran en el encéfalo y la médula espinal. Los gliomas se clasifican según las características histológicas y moleculares, y representan el tipo más habitual de tumor del sistema nervioso central (SNC) en los niños.
Históricamente, los gliomas en pediatría se clasificaban en gliomas de grado bajo (grados 1–2 de la Organización Mundial de la Salud [OMS]) y de grado alto (grados 3–4 de la OMS) según sus características histológicas. Sin embargo, la incorporación de biomarcadores moleculares ha dado lugar a un nuevo esquema de clasificación. De acuerdo con la clasificación de tumores del sistema nervioso central de la OMS de 2021 (5ª edición), los gliomas, los tumores glioneuronales y los tumores neuronales se clasifican en general en gliomas difusos de tipo adulto, gliomas difusos de grado bajo de tipo pediátrico, gliomas difusos de grado alto de tipo pediátrico, gliomas astrocíticos circunscritos, tumores glioneuronales y neuronales y tumores ependimarios.[
Los resúmenes del PDQ sobre tratamiento de los tumores encefálicos infantiles se organizan principalmente de acuerdo con la clasificación de tumores del SNC de la OMS de 2021.[
Características anatómicas
Los gliomas infantiles aparecen en cualquier parte del SNC (consultar la Figura 1). Para conocer la ubicación más frecuente en el SNC de cada tipo de tumor, consultar el Cuadro 2. 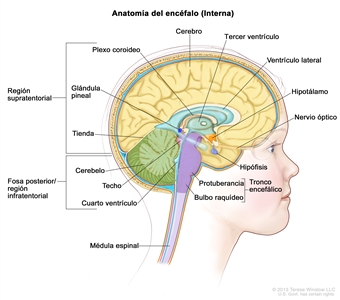
Características clínicas
Los síntomas de presentación inicial de los gliomas infantiles dependen de los siguientes aspectos:
- Ubicación anatómica.
- Tamaño del tumor.
- Tasa de crecimiento tumoral.
- Edad cronológica y de desarrollo del niño.
Los lactantes y niños pequeños con gliomas circunscritos (con mayor frecuencia, astrocitomas pilocíticos) y, con menor frecuencia, astrocitomas difusos que comprometen el hipotálamo, tal vez presenten síndrome diencefálico, que se manifiesta por el retraso del crecimiento en un niño demacrado y aparentemente eufórico. Es posible que estos niños presenten pocos hallazgos neurológicos, pero quizás presenten macrocefalia, letargo intermitente o deterioro visual.[
A veces, los niños con gliomas difusos de línea media centrados en la protuberancia (antes llamados gliomas pontinos intrínsecos difusos [GPID]) presentan la siguiente tríada clásica de síntomas; sin embargo, en ocasiones se presentan solo uno o dos de estos síntomas en el momento del diagnóstico:
- Neuropatías craneales, en especial paresia del nervio motor ocular externo.
- Signos de tractos largos.
- Ataxia.
La hidrocefalia obstructiva debida a la expansión de la protuberancia también es un síntoma de presentación inicial en algunos casos. Además, es posible que se presenten síntomas inespecíficos, como cambios conductuales o disminución del rendimiento escolar.
La presentación clínica de los astrocitomas circunscritos (por ejemplo, astrocitomas pilocíticos) en el tronco encefálico depende de la localización del tumor. Algunos de los síntomas de presentación inicial habituales son los siguientes:[
- Presión intracraneal elevada con hidrocefalia.
- Hemiparesia unilateral.
- Neuropatías craneales unilaterales.
- Ataxia.
Evaluación diagnóstica
La evaluación diagnóstica inicial de los pacientes con gliomas incluye imágenes por resonancia magnética (IRM) del encéfalo o la columna vertebral, con contraste o sin este. El riesgo de diseminación por el eje encefalomedular depende del tipo tumoral y se pueden tomar imágenes completas del eje encefalomedular, incluso IRM del encéfalo y la columna vertebral completa, en pacientes seleccionados. En la mayoría de los casos, el diagnóstico específico se determina después de la intervención quirúrgica y la clasificación patológica.
Los tumores primarios del tronco encefálico se diagnostican con mayor frecuencia en función de los hallazgos clínicos y los estudios de neuroimágenes mediante IRM, de las siguientes maneras:[
- Glioma difuso de línea media centrado en la protuberancia (DIPG). De manera sistemática, cuando no se cuenta con un diagnóstico histológico, se emplea un diagnóstico presunto de DIPG conforme a características clásicas en las imágenes y características clínicas. Sin embargo, cada vez más se obtiene la confirmación histológica para el ingreso en estudios de investigación y para la caracterización molecular del tumor.[
6 ] Debido a los desafíos técnicos de las biopsias de la protuberancia, es preferible que un neurocirujano pediátrico experimentado realice el procedimiento con el fin de reducir el riesgo de complicaciones neurológicas irreversibles.[7 ,8 ,9 ,10 ,11 ] Se recomienda la biopsia para los tumores pontinos cuando no se obtiene un diagnóstico definitivo con los resultados de las imágenes. - Tumores de tronco encefálico diferentes a DIPG. Por lo general, se indica biopsia o resección para los tumores de tronco encefálico diferentes a DIPG.
No se suele realizar la punción lumbar para examinar el líquido cefalorraquídeo en busca de células tumorales circulantes en los niños que presentan estos tipos de tumor.
Clasificación de la Organización Mundial de la Salud de los gliomas, los astrocitomas, y los tumores glioneuronales o neuronales
La clasificación patológica de los tumores encefálicos infantiles es un área de gran especialización que sigue en desarrollo. Los avances rápidos en la genética molecular han producido mejoras importantes en el diagnóstico exacto de los tumores encefálicos en la última década. Al mismo tiempo, se han identificado muchas entidades tumorales nuevas en el encéfalo a partir de características moleculares únicas. Es muy recomendable que un especialista en neuropatología experimentado examine el tejido diagnóstico, junto con las pruebas moleculares, si estuvieran disponibles.
De acuerdo con la clasificación de los tumores del SNC de la OMS de 2021, los gliomas y los tumores glioneuronales o neuronales que se presentan de manera predominante en la infancia se clasifican de la siguiente manera:
- Gliomas difusos de grado alto de tipo pediátrico.
- Gliomas difusos de grado bajo de tipo pediátrico.
- Gliomas astrocíticos circunscritos.
- Tumores glioneuronales y neuronales.
- Tumores ependimarios. Para obtener más información, consultar Tratamiento del ependimoma infantil.
Dentro de cada tipo de tumor, se distinguen varios subtipos según las características histológicas y moleculares.
En la clasificación de los tumores del SNC de la OMS de 2021 se recomienda la siguiente estructura de informes por niveles:[
- Diagnóstico integrado (diagnóstico histológico y molecular combinado basado en tejidos).
- Diagnóstico histológico.
- Grado de la OMS para los tumores del SNC.
- Información molecular (enumerada).
Clasificación por grados de la Organización Mundial de la Salud para los tumores del sistema nervioso central
Mientras que los tumores del SNC se clasificaron con anterioridad solo en función de sus características histopatológicas y comportamiento clínico (clasificación clinicopatológica), el esquema de clasificación por grados de la OMS de 2021 para los tumores del SNC emplea una clasificación histológica y molecular combinada para muchos tipos de tumores.[
En el Cuadro 1 se muestra la clasificación y los grados de la OMS de 2021 para los tipos y subtipos más frecuentes de gliomas, tumores glioneuronales y tumores neuronales (salvo los tumores ependimarios) que se presentan en el SNC durante la infancia y la adolescencia.
| Tipo o subtipo de tumor | Grados de la OMS para los tumores del SNC | |
|---|---|---|
| Gliomas difusos de grado alto de tipo pediátrico: | ||
| Glioma difuso de línea media con alteración H3 K27 | 4 | |
| Glioma difuso de grado alto de tipo pediátrico con H3 natural eIDHnatural | 4 | |
| Glioma hemisférico de tipo infantil (de lactantes) | Sin asignación | |
| Gliomas difusos de grado bajo de tipo pediátrico: | ||
| Glioma difuso de grado bajo con alteración de la vía MAPK | Sin asignación | |
| Astrocitoma difuso con alteración deMYBoMYBL1 | 1 | |
| Gliomas astrocíticos circunscritos: | ||
| Astrocitoma pilocítico | 1 | |
| Astrocitoma de grado alto con características piloides | Sin asignación | |
| Xantoastrocitoma pleomórfico | 2, 3 | |
| Astrocitoma subependimario de células gigantes | 1 | |
| Tumores glioneuronales y neuronales: | ||
| Ganglioglioma | 1 | |
| Ganglioglioma desmoplásico infantil o astrocitoma desmoplásico infantil (de lactantes) | 1 | |
| Tumor neuroepitelial disembrioplásico | 1 | |
Ubicación en el sistema nervioso central
Los gliomas infantiles se presentan en cualquier parte del SNC, aunque cada tipo de tumor suele hacerlo en ubicaciones anatómicas específicas (consultar el Cuadro 2).
| Tipo de tumor | Ubicación frecuente en el sistema nervioso central |
|---|---|
| Gliomas astrocíticos circunscritos | Cerebelo, nervio óptico, quiasma óptico o hipotálamo, tálamo y ganglios basales, tronco encefálico, hemisferios cerebrales y médula espinal (infrecuente) |
| Ganglioglioma | Cerebro, tronco encefálico; en ocasiones, otras ubicaciones |
| Glioma difuso de línea media con alteración H3 K27 | Protuberancia, tálamo, médula espinal y otras estructuras de la línea media |
| Glioma difuso de grado alto de tipo pediátrico con H3 natural eIDHnatural | Cerebro; en ocasiones, otras ubicaciones |
Cerebelo: más del 80 % de los gliomas ubicados en el cerebelo son astrocitomas pilocíticos (grado I de la OMS) y, con frecuencia, quísticos; la mayoría de los restantes son gliomas difusos de grado bajo de tipo pediátrico.[
Tronco encefálico: el término glioma del tronco encefálico es una descripción genérica que hace referencia a cualquier tumor de origen neuroglial que surja en el tronco encefálico, incluso el mesencéfalo, la protuberancia y el bulbo raquídeo. Si bien se presentan otros tipos histológicos (por ejemplo, ganglioglioma) en el tronco encefálico, predominan los dos siguientes:
- Glioma difuso de línea media con alteración H3 K27 centrado en la protuberancia.[
13 ] Estos se han llamado habitualmente gliomas pontinos intrínsecos difusos (GPID) debido a su ubicación anatómica. Para obtener más información sobre el glioma difuso de línea media con alteración H3 K27, consultar la sección Características genómicas de los gliomas, los tumores glioneuronales y los tumores neuronales. - Astrocitomas pilocíticos, que se presentan en todo el tronco encefálico.
Los tumores con componentes exofíticos son, en su mayoría, astrocitomas pilocíticos.[
Vía óptica e hipotálamo: la mayoría de los tumores que surgen dentro de la vía óptica (es decir, nervio óptico, quiasma y radiaciones ópticas) representan astrocitomas pilocíticos y, en raras ocasiones, gliomas difusos de grado bajo de tipo pediátrico.[
Cerebro: la mayoría de los tumores que surgen en los hemisferios cerebrales comprenden gliomas astrocíticos circunscritos y gliomas difusos de grado bajo de tipo pediátrico, seguidos de gliomas difusos de grado alto de tipo pediátrico.[
Características genómicas de los gliomas, los tumores glioneuronales y los tumores neuronales
Síndromes de susceptibilidad al cáncer seleccionados relacionados con los gliomas infantiles
Neurofibromatosis de tipo 1
Los niños con neurofibromatosis de tipo 1 (NF1) tienen una mayor propensión a presentar gliomas de grado bajo, en especial en la vía óptica. Hasta el 20 % de los pacientes con NF1 presentarán un glioma de la vía óptica. La mayoría de los niños con gliomas del nervio óptico relacionados con la NF1 son asintomáticos o tienen síntomas no progresivos y no requieren tratamiento antitumoral. Por lo general, no está indicada la detección sistemática con imágenes por resonancia magnética (IRM) en pacientes asintomáticos con NF1, aunque algunos investigadores obtienen una IRM al inicio para niños pequeños que no se pueden someter a exámenes oftalmológicos detallados.[
El diagnóstico a menudo se basa en los hallazgos clínicos compatibles y las características de las imágenes. La confirmación histológica casi nunca es necesaria en el momento del diagnóstico. Cuando se realizan biopsias, los tumores que se encuentran son de manera predominante astrocitomas pilocíticos.[
Las indicaciones para el tratamiento varían y, a menudo, se basan en el objetivo de preservar la visión.
En muy pocas ocasiones, los pacientes con NF1 presentan gliomas de grado alto. A veces, estos tumores son el resultado de una transformación de un tumor de grado inferior.[
Esclerosis tuberosa
Los pacientes con esclerosis tuberosa son más propensos a presentar astrocitoma subependimario de células gigantes (SEGA). Las variantes de TSC1 o TSC2 causan la activación constitutiva de la vía de señalización del complejo 1 del blanco de la rapamicina en los mamíferos (mTORC1), lo que aumenta la proliferación. Los SEGA responden a los abordajes molecularmente dirigidos con inhibidores de la vía mTORC1.[
Características moleculares y alteraciones genómicas recurrentes
Las alteraciones genómicas recurrentes que provocan la activación constitutiva de la vía de la proteína cinasa activada por mitógenos (MAPK), que con mayor frecuencia afecta al gen BRAF, representan el principal (y a menudo único) iniciador oncogénico en la gran mayoría de los gliomas infantiles de grado bajo, como los astrocitomas pilocíticos o pilomixoides, los gangliogliomas y otros.[
Los genomas tumorales más complejos son característicos de los gliomas difusos de grado alto de tipo pediátrico. Estos genomas complejos incluyen alteraciones genómicas recurrentes en genes codificadores de histonas H3 (por ejemplo, H3F3A, HIST1H3B), genes de las vías de reparación del daño al DNA (por ejemplo, TP53, PPM1D, ATM, MDM2), genes modificadores de la cromatina (por ejemplo, ATRX, BCOR, SETD2), genes de las vías del ciclo celular (por ejemplo, CDKN2A, CDKN2B, RB1) o presentan amplificaciones oncogénicas (PDGFR, VEGFR2, KIT, MYC, MYCN).[
Un subconjunto infrecuente de gliomas de grado alto de tipo pediátrico que surgen en pacientes con deficiencia hereditaria y bialélica en la reparación de los errores de emparejamiento (bMMRD) se caracteriza por una carga mutacional extremadamente alta. La identificación correcta de estos pacientes en el momento del diagnóstico es fundamental debido a la resistencia intrínseca a la temozolomida y la sensibilidad al tratamiento con inhibidores de puntos de control inmunitario.[
BRAF::KIAA1549
En el astrocitoma pilocítico, la activación de BRAF sucede con mayor frecuencia por una fusión génica BRAF::KIAA1549, que genera una proteína de fusión sin el dominio autorregulador de BRAF.[
La presencia de la fusión BRAF::KIAA1549 se relaciona con una mejora del desenlace clínico (supervivencia sin progresión [SSP] y supervivencia general [SG]) en pacientes con astrocitoma pilocítico.[
Variantes deBRAF
Las variantes puntuales activadoras de BRAF, con mayor frecuencia BRAF V600E, están presentes en un subconjunto de gliomas infantiles y tumores glioneuronales que abarcan varios tipos histológicos, como el xantoastrocitoma pleomórfico, el astrocitoma pilocítico, el ganglioglioma, el astrocitoma o el ganglioglioma desmoplásico infantil (de lactantes), entre otros.[
En estudios clínicos retrospectivos se observó lo siguiente:
- En una serie retrospectiva de más de 400 niños con gliomas de grado bajo, el 17 % de los tumores exhibían una variante BRAF V600E. La tasa de SSP a 10 años fue del 27 % en los pacientes que tenían tumores con variante BRAF V600E, en comparación con el 60 % en los pacientes cuyos tumores no albergaban esa variante. Otros factores relacionados con este pronóstico precario fueron la resección subtotal y la deleción de CDKN2A.[
26 ][Nivel de evidencia C2] Incluso en los pacientes sometidos a una resección macroscópica total, se observó recidiva en un tercio de ellos, lo que indica que los tumores que tienen la variante BRAF V600E tienen un fenotipo más invasivo que otras variantes de glioma de grado bajo. - En un análisis similar, los niños con astrocitomas diencefálicos de grado bajo con una variante BRAF V600E tuvieron una tasa de SSP a 5 años del 22 %, en comparación con una tasa de SSP del 52 % en los niños con BRAF natural.[
27 ][Nivel de evidencia C2] - La frecuencia de la variante BRAF V600E en casos pediátricos fue significativamente superior en los gliomas de grado bajo que se transformaron en gliomas de grado alto (8 de 18 casos), en comparación con la frecuencia de la variante en los casos sin transformación (10 de 167 casos).[
24 ]
Variantes deNF1
Las alteraciones somáticas en NF1 se observan con mayor frecuencia en niños con NF1 y se relacionan con alteraciones de la línea germinal en el supresor tumoral NF1. La pérdida de heterocigosis para NF1 representa la alteración somática más común en estos pacientes seguida de variantes inactivadoras del segundo alelo de NF1, y es compatible con la lesión secundaria ("segundo golpe") necesaria para la carcinogénesis. Si bien la mayoría de los pacientes con gliomas de grado bajo que presentan alteraciones en NF1 tienen un pronóstico excelente a largo plazo, es posible que se produzca una transformación secundaria a un glioma de grado alto en un subgrupo pequeño de estos casos. Desde el punto de vista genómico, la transformación se vincula con la adquisición de otros iniciadores oncogénicos, como alteraciones funcionales de pérdida de función en CDKN2A, CDKN2B o ATRX. Los pacientes con NF1 también presentan gliomas primarios de grado alto, pero esto es muy infrecuente. Las alteraciones genómicas fuera de NF1 que afectan la vía de señalización MAPK son muy infrecuentes en los gliomas que se presentan en niños con NF1.[
Fusiones de los genesALK,NTRK1,NTRK2,NTRK3oROS1
Los gliomas de grado alto con características moleculares típicas surgen en lactantes, por lo general se diagnostican durante el primer año de vida.[
Las fusiones génicas de ROS1 también se han notificado en los gliomas que se presentan en niños de más edad y en adultos. En un metanálisis retrospectivo, en el que se incluyeron 40 niños mayores de 1 año, se encontró que las fusiones génicas de ROS1 se presentaron en gliomas de diferentes tipos histológicos, incluso en gliomas difusos de grado alto y de grado bajo, así como en tumores glioneuronales.[
Otras alteraciones genómicas
Además de la activación de BRAF o la pérdida de NF1, se han observado otras alteraciones primarias iniciadoras oncogénicas en la vía de señalización MAPK en astrocitomas pilocíticos y otros gliomas de tipo pediátrico. Estos incluyen variantes oncogénicas o fusiones que afectan FGFR1, FGFR2, PTPN11, RAF1, NTRK2 y otros genes.[
Los gliomas de grado bajo con reordenamientos en la familia de factores de transcripción MYB [
Gliomas angiocéntricos
Los gliomas angiocéntricos habitualmente surgen en niños y adultos jóvenes como tumores encefálicos que causan convulsiones.[
En dos informes de 2016 se identificaron alteraciones en el gen MYB presentes en casi todos los casos diagnosticados como gliomas angiocéntricos; el gen QKI fue el principal compañero de fusión en los casos en los que fue posible obtener pruebas sobre la pareja de fusión.[
Astroblastomas con alteración deMN1
Los astroblastomas se definen según sus características histológicas como neoplasias gliales compuestas de células positivas para GFAP que contienen pseudorosetas astroblásticas, a menudo con esclerosis. Los astroblastomas se diagnostican de manera principal durante la niñez y hasta el comienzo de la edad adulta.[
En los siguientes estudios se caracterizaron las alteraciones genómicas asociadas con el astroblastoma:
- En un informe se detalló una clasificación molecular de los tumores neuroectodérmicos primitivos (TNEP) del SNC y se descubrió una entidad nueva llamada tumor neuroepitelial de grado alto del SNC con alteración de MN1 (CNS HGNET-MN1) que se caracterizó por la presencia de fusiones génicas que afectan el gen MN1.[
39 ] La mayoría de los tumores con diagnóstico histológico de astroblastoma (16 de 23) pertenecían a esta entidad definida por sus características moleculares. - En un informe de 27 casos de astroblastomas definidos por sus características histológicas se encontraron 10 casos con reordenamientos de MN1, 7 casos con reordenamientos de BRAF y 2 casos con reordenamientos de RELA.[
40 ] Con el análisis de matriz de metilación se observó que los casos con reordenamientos de MN1 se agruparon con el CNS HGNET-MN1, los casos con alteraciones de BRAF se agruparon con los xantoastrocitomas pleomórficos, y los casos con alteración en RELA se agruparon con los ependimomas. - En la evaluación genómica de 8 casos de astroblastomas se detectaron 4 casos con alteraciones de MN1. De los 4 casos restantes, 2 presentaron alteraciones genómicas compatibles con glioma de grado alto y 2 casos no se pudieron clasificar según sus características moleculares.[
41 ] - En un estudio se caracterizaron 8 casos de astroblastoma. Los 5 casos sometidos a análisis de hibridación fluorescente in situ mostraron reordenamientos de MN1.[
42 ]
En estos informes se indica que el diagnóstico histológico del astroblastoma abarca un grupo heterogéneo de entidades definidas por sus características genómicas. Los astroblastomas con fusiones de MN1 representan un subconjunto diferenciado de los casos diagnosticados por sus características histológicas.[
Variantes deIDH1eIDH2
Los tumores con alteraciones de IDH1 e IDH2 ocurren en la población pediátrica como los gliomas de grado bajo (grado 2 de la OMS), los gliomas de grado alto (grado 3 y 4 de la OMS), y los oligodendrogliomas con codeleción de 1p y 19q. Para obtener más información sobre los gliomas con alteración en IDH1 y IDH2, consultar la sección variantes de IDH1 e IDH2 en Características moleculares de los gliomas de grado alto de tipo pediátrico.
Características moleculares de los gliomas de grado alto de tipo pediátrico
Los gliomas infantiles de grado alto son diferentes, desde el punto de vista biológico, de los que surgen en adultos.[
Subgrupos definidos mediante los patrones de metilación del DNA
Los gliomas de grado alto de tipo pediátrico se pueden separar en subgrupos característicos a partir de patrones epigenéticos (metilación del DNA). Estos subgrupos exhiben ganancias o pérdidas del número de copias cromosómicas y variantes génicas en el tumor que son características.[
Los siguientes subgrupos de glioma de grado alto de tipo pediátrico se identificaron a partir de sus patrones de metilación del DNA y muestran características clínicas y moleculares distintivas:[
Alteraciones genómicas asociadas con los gliomas difusos de línea media
Variantes de histonas en K27: variantes de H3.3 (H3F3A) y H3.1 (HIST1H3By, con menor frecuencia,HIST1H3C) en K27 y variantes de EZHIP
Los casos con alteraciones de histonas en K27 se presentan sobre todo en la mitad de la niñez (mediana de edad, alrededor de 10 años), surgen casi exclusivamente en la línea media (tálamo, tronco encefálico y médula espinal) y acarrean un pronóstico muy precario. En la clasificación de la OMS de 2021 se agrupan estos cánceres en una sola entidad: glioma difuso de línea media con alteración H3 K27. Sin embargo, hay diferencias clínicas y biológicas entre los casos con variantes de H3.3 y H3.1, como se describe a continuación.[
Los casos de glioma difuso de línea media con alteración H3 K27 se definen por la pérdida de la trimetilación de H3 K27 debido a una variante H3 K27M, o con menor frecuencia, por sobreexpresión de EZHIP. Esta entidad incluye la mayoría de los gliomas de grado alto del hipotálamo, la protuberancia (gliomas pontinos intrínsecos difusos [GPID]) y la médula espinal, de predominio en niños, pero que también se presenta en adultos.[
Gliomas con H3.3 K27M: estos casos surgen por toda la línea media y la protuberancia; corresponden a cerca del 60 % de los casos en estos sitios y, por lo común, aparecen entre los 5 y 10 años de edad.[
Gliomas con H3.1 K27M: los casos con la alteración H3.1 K27M son casi 5 veces menos comunes que los casos con la alteración H3.3 K27M. Surgen de manera primaria en la protuberancia y se presentan a una edad más temprana que los otros casos del subtipo H3.3 K27M (mediana de edad, 5 vs. 6–10 años). Estos pacientes tienen un pronóstico algo más favorable que los casos del subtipo H3.3 K27M (mediana de supervivencia, 15 vs. 11 meses). Las variantes de ACVR1, que también es la variante observada en la afección genética fibrodisplasia osificante progresiva, están presentes en una proporción alta de casos con la alteración H3.1 K27M.[
Gliomas con H3.2 K27M: en escasas ocasiones, también se han identificado variantes K27M en casos con alteración en H3.2 (HIST2H3C).[
Un subgrupo de tumores con variantes H3 K27 presentarán al mismo tiempo una variante BRAF V600E o una variante de FGFR1. En una cohorte retrospectiva de 29 tumores, que se combinó con 31 casos que se habían publicado en el pasado, se demostró una predisposición mayor por la ubicación en el tálamo. Estos casos exhibían un grupo de metilación del DNA singular que se diferencia de otros subgrupos de glioma difusos de línea media y subtipos de glioma con alteraciones de BRAF o FGFR1. La mediana de supervivencia de estos pacientes superó los 3 años.[
Sobrexpresión de EZHIP: la pequeña minoría de pacientes con gliomas difusos de línea media que carecen de variantes de la histona H3 a menudo exhiben sobrexpresión de EZHIP.[
Variante de H3.3 (H3F3A) en G34
Los subtipos con alteración H3.3 G34 se presentan por variantes de H3.3 cuando cambia la glicina en posición 34 por arginina o valina (G34R/V).[
Los pacientes con variantes de H3F3A tienen un riesgo alto de fracaso del tratamiento,[
Variantes deIDH1eIDH2
Los tumores con alteraciones de IDH1 y de IDH2 se presentan en la población pediátrica como gliomas de grado bajo (grado 2 de la OMS), gliomas de grado alto (grados 3 y 4 de la OMS) y oligodendrogliomas con codeleción de 1p y 19q.[
- Las variantes de IDH1 son mucho más comunes que las variantes de IDH2 y se encuentran en alrededor del 90 % de los tumores del SNC con alteración de IDH.
- Los gliomas de grado bajo con alteración de IDH son más comunes que los gliomas de grado alto con alteración de IDH, y representan alrededor de tres cuartas partes de los casos de gliomas pediátricos con alteración de IDH.
- Los oligodendrogliomas con variantes de IDH representan cerca del 20 % de los tumores pediátricos en el SNC con variantes de IDH.
- La mediana de edad en el momento del diagnóstico para los pacientes pediátricos con tumores que exhiben alteración de IDH es de alrededor de 16 años, y los tumores en el SNC con alteración de IDH son muy poco frecuentes en niños de 10 años o menos.
- Al igual que en los astrocitomas con variantes de IDH que se presentan en los adultos, los que se observan en los niños con frecuencia exhiben variantes de TP53 (cerca del 90 % de los casos) y variantes de ATRX (cerca del 50 % de los casos).
- Del mismo modo que los gliomas de grado bajo con alteración de IDH, los tumores de grado bajo en pacientes pediátricos pueden progresar y convertirse en gliomas de grado alto.
Los casos con alteraciones de IDH1 representan una proporción baja de los gliomas de grado alto (cerca del 5–10 %) que se observan en el ámbito pediátrico. La mayoría de los casos corresponden a adolescentes mayores (mediana de edad en una población pediátrica, 16 años) con tumores hemisféricos.[
Los pacientes pediátricos con variantes de IDH1 tienen un pronóstico más favorable que aquellos con otros tipos de gliomas de grado alto.[
Se han notificado gliomas de grado alto raros, con alteración de IDH en niños con síndromes de deficiencia en la reparación de errores de emparejamiento (síndrome de Lynch o síndrome de deficiencia constitucional en la reparación de errores de emparejamiento).[
Tumor similar al xantoastrocitoma pleomórfico
Alrededor del 10 % de los gliomas infantiles de grado alto exhiben patrones de metilación del DNA que son similares a los del xantoastrocitoma pleomórfico (XAP).[
Astrocitoma de grado alto con características piloides
Esta entidad se incluyó en la clasificación de la OMS de 2016 (llamada astrocitoma pilocítico con anaplasia) para describir los tumores con características histológicas del astrocitoma pilocítico, aumento de la actividad mitótica y características adicionales de grado alto. La nomenclatura actual se adoptó en la clasificación de la OMS de 2021. En una publicación más reciente se describió una cohorte de 83 casos con estas características histológicas (tumores denominados astrocitomas anaplásicos con características piloides) que compartían un perfil de metilación del DNA común, distinto de los perfiles de metilación de otros gliomas. Estos tumores se presentaron con más frecuencia en adultos (mediana de edad, 41 años) y con frecuencia albergaban deleciones de CDKN2A/B, alteraciones en la vía MAPK (más a menudo en el gen NF1) y variantes o deleciones de ATRX. Se relacionan con una evolución clínica intermedia entre el astrocitoma pilocítico y el glioblastoma con IDH natural.[
Otras variantes
Los pacientes pediátricos con glioma de grado alto tipo glioblastoma multiforme cuyos tumores carecen de variantes de histonas y variantes de IDH1 representan alrededor del 40 % de los casos de glioblastoma multiforme infantil.[
Gliomas de grado alto en lactantes
Los lactantes y niños pequeños con gliomas de grado alto tienen tumores con características moleculares típicas [
En dos estudios de las características moleculares de los gliomas de grado alto en lactantes y niños pequeños se definió la naturaleza distintiva de los tumores que surgen en niños menores de 1 año. Un hallazgo clave de ambos estudios es la importancia de las fusiones de genes relacionados con tirosina–cinasas (por ejemplo, ALK, NTRK1, NTRK2, NTRK3 y ROS1) en pacientes de este grupo de edad. En ambos estudios también se encontró que los lactantes con gliomas de grado alto cuyos tumores tienen estas fusiones génicas tienen tasas de supervivencia mucho más altas que las de los niños mayores con gliomas de grado alto.[
En el primer estudio se presentaron datos de 118 niños menores de 1 año con diagnóstico de glioma de grado bajo o alto que tenían tejido tumoral disponible para la caracterización genómica.[
- Los tumores del grupo 1 están determinados por receptores tirosina–cinasas (RTK) y en su mayoría son de grado alto (83 %). Estos tumores albergan lesiones en ALK, ROS1, NTRK y MET. La mediana de edad en el momento del diagnóstico es de 3 meses, y las tasas de SG se acercan al 60 %.
- Los tumores del grupo 2 están determinados por RAS/MAPK; todos son gliomas hemisféricos de grado bajo, lo que representa un cuarto de los gliomas hemisféricos en lactantes. La alteración más común es la variante BRAF V600E, seguida de alteraciones en FGFR1 y fusiones de BRAF. En este grupo, la mediana de edad en el momento de la presentación inicial es de 8 meses y su pronóstico es el más favorable de todos (tasa de SG a 10 años del 93 %).
- Los tumores del grupo 3 están determinados por RAS/MAPK, su tipo histológico es de grado bajo y el sitio de origen es la línea media (alrededor del 80 % son gliomas de vía óptica y de hipotálamo). La mayoría de los tumores del grupo 3 exhiben fusiones de BRAF o alteraciones BRAF V600E. La mediana de edad en el momento del diagnóstico es de 7,5 meses. La tasa de supervivencia sin progresión (SSP) a los 5 años fue de cerca del 20 %, y la tasa de SG a los 10 años fue de alrededor del 50 % (muy inferior a la tasa de los gliomas de vía óptica o hipotálamo en niños >1 año).
El segundo estudio se centró en los tumores de niños menores de 4 años con un diagnóstico patológico de gliomas, astrocitomas o tumores glioneuronales de grado 2, 3 y 4 de la OMS. Entre los 191 tumores estudiados que cumplían los criterios de inclusión, 61 tenían perfiles de metilación compatibles con los subtipos de glioma que se presentan en niños mayores (por ejemplo, IDH1, glioma difuso de línea media con alteración H3 K27, SEGA, xantoastrocitoma pleomórfico, etc.). Los 130 casos restantes se denominaron conjunto intrínseco y fueron objeto de caracterización molecular adicional:[
- El conjunto intrínseco contenía a la mayoría de los pacientes diagnosticados antes de tener 1 año de edad (49 de 63 pacientes, 78 %) que tenían una mediana de edad de 7,2 meses. Con frecuencia, los tumores se encontraban en una ubicación hemisférica superficial, a menudo con compromiso de las meninges, pero con un borde bien definido con el encéfalo normal adyacente.
- El clasificador de metilación colocó la mayoría de estos casos en el subgrupo de ganglioglioma desmoplásico infantil o astrocitoma (DIG/DIA) o en el subgrupo de glioma hemisférico infantil.
- En el conjunto intrínseco, se disponía de tejido para la secuenciación del panel génico y del ARN de 41 tumores, entre ellos, 25 tumores presentaron fusiones que afectaban ALK (n = 10), NTRK1 (n = 2), NTRK2 (n = 2) , NTRK3 (n = 8 ), ROS1 (n = 2) o MET (n = 1). Se observaron variantes de BRAF (n = 3) en los casos con puntuación alta en la matriz de metilación para los subgrupos DIG/DIA o similar a DIG/DIA.
- En los pacientes del grupo intrínseco, la tasa de supervivencia a 5 años fue más alta para los pacientes cuyos tumores tenían fusiones génicas, en comparación con los pacientes cuyos tumores no tenían fusiones (80 vs. 60 %, respectivamente). Sin embargo, ambos grupos de pacientes tuvieron tasas de supervivencia mucho más altas que otros niños con gliomas de grado alto.
Gliomas de grado alto secundarios
Los gliomas infantiles de grado alto secundarios (glioma de grado alto precedido por un glioma de grado bajo) son poco comunes (2,9 % en un estudio de 886 pacientes). Ningún glioma infantil de grado bajo con la fusión BRAF::KIAA1549 se transformó en un glioma de grado alto, mientras que los gliomas de grado bajo con variantes BRAF V600E se relacionaron con un aumento del riesgo de transformación. De los 18 pacientes con glioma secundario de grado alto, 7 (aproximadamente el 40 %) presentaron variantes BRAF V600E, y 8 de 14 casos (57 %) presentaron alteraciones en CDKN2A.[
Características moleculares de los tumores glioneuronales y neuronales
Los tumores glioneuronales y los tumores neuronales por lo general son tumores de grado bajo. Algunos tipos histológicos reconocidos por la clasificación de la OMS de 2021 son los siguientes:[
- Ganglioglioma.
- Ganglioglioma desmoplásico infantil o astrocitoma desmoplásico infantil (de lactantes).
- Tumor neuroepitelial disembrioplásico.
- Tumor glioneuronal papilar.
- Tumor glioneuronal formador de rosetas.
- Gangliocitoma displásico cerebeloso (enfermedad de Lhermitte-Duclos).
- Gangliocitoma.
- Tumor glioneuronal leptomeníngeo difuso.
- Neurocitoma central.
- Neurocitoma extraventricular.
Ganglioglioma
El ganglioglioma se presenta en niños y adultos. Produce convulsiones y aparece con más frecuencia en la corteza cerebral, pero a veces surge en otros sitios, como la médula espinal.[
El elemento fundamental de la patogénesis molecular del ganglioglioma son las alteraciones genómicas que conducen a la activación de la vía MAPK.[
Astrocitoma desmoplásico infantil y ganglioglioma desmoplásico infantil
Los astrocitomas desmoplásicos infantiles (DIA) y los gangliogliomas desmoplásicos infantiles (DIG) se presentan con mayor frecuencia en el primer año de vida, es decir en lactantes, y muestran una apariencia característica en las imágenes donde se ve un nódulo sólido que se realza con el contraste y se acompaña de un componente quístico grande.[
Las alteraciones genómicas observadas con mayor frecuencia en el DIA y el DIG son variantes de BRAF que afectan V600. Las fusiones génicas que afectan genes de cinasas se observan con menos frecuencia.
- Entre 16 casos de DIA y DIG confirmados mediante análisis histológico y de perfil de metilación del DNA, se identificaron variantes de BRAF en 7 casos (43,8 %): 4 variantes BRAF V600E y 3 variantes BRAF V600D.[
69 ] Otro caso tenía una fusión EML4::ALK. Se presentaron variantes de BRAF en 4 de 12 (25 %) casos de DIG (3 de 4 casos con la alteración BRAF V600D) y en 3 de 4 (75 %) casos de DIA (3 casos con la alteración BRAF V600E). - En un estudio de 7 casos de DIG se identificaron alteraciones en la vía MAPK en 4 casos (57 %).[
70 ] Entre ellas, 3 alteraciones que afectaban el gen BRAF (V600E, V600D y una deleción o inserción en V600) y otra se trató de una fusión en el marco de lectura TPM3::NTRK1. Cabe aclarar que la frecuencia de variantes alélicas fue baja (8–27 %), lo que indica que el DIG se caracteriza por un componente no neoplásico abundante que se traduce en frecuencias bajas de alelos con variantes clonales oncoiniciadoras. - En otro informe también se identificó la variante BRAF V600D en un caso de DIG.[
71 ] Debido a que la variante V600D es mucho menos frecuente que la variante V600E en otros tipos de cáncer, la detección de esta variante en varios casos de DIG indica una relación de la variante con esta enfermedad.
Tumor neuroepitelial disembrioplásico
El tumor neuroepitelial disembrioplásico (TNED) se presenta en niños y adultos con una mediana de edad en el momento del diagnóstico entre la adolescencia media o tardía. Desde el punto histopatológico, se caracteriza por filas de células de apariencia oligodendroglial y células ganglionares corticales flotando en mucina.[
Se han notificado alteraciones en FGFR1 en el 60 % a 80 % de los TNED, entre ellas variantes puntuales activadoras de FGFR1, duplicaciones internas en tándem del dominio cinasa y fusiones génicas activadoras.[
Tumor glioneuronal papilar
El tumor glioneuronal papilar es una neoplasia bifásica de grado bajo con diferenciación astrocítica y neuronal que casi siempre aparece en el compartimiento supratentorial.[
La alteración genómica principal relacionada con el tumor glioneuronal papilar es una fusión génica, SLC44A1::PRKCA, que se relaciona con la translocación t(9:17)(q31;q24).[
Tumor glioneuronal formador de rosetas
El tumor glioneuronal formador de rosetas (TGNR) se presenta en adolescentes y adultos, a menudo se ubica a nivel infratentorial, pero también surge en las regiones mesencefálica y diencefálica.[
El perfil de metilación del DNA indica que el TGNR exhibe un perfil epigenético propio que lo diferencia de otras entidades tumorales glioneuronales o neurogliales de grado bajo.[
Tumor glioneuronal leptomeníngeo difuso
El tumor glioneuronal leptomeníngeo difuso (DLGNT) es un tipo raro de tumor del SNC que se caracteriza desde el punto de vista radiográfico por el realce leptomeníngeo en la IRM. Este tumor suele afectar la fosa posterior, la región del tronco encefálico y la médula espinal.[
El DLGNT exhibió un perfil epigenético específico en las matrices de metilación del DNA, y la agrupación no supervisada de datos de matrices aplicadas en 30 casos permitió determinar dos subtipos según el tipo de metilación: MC-1 (n = 17) y MC-2 (n = 13).[
- Los 30 casos exhibieron pérdida del cromosoma 1p, pero solo 6 de 17 casos de DLGNT-MC-1 presentaron ganancia adicional del cromosoma 1q, en comparación con todos los casos de DLGNT-MC-2.[
80 ] En otro informe se encontró que la ganancia del cromosoma 1q fue un factor de pronóstico adverso en pacientes con DLGNT (incluso casos con enfermedad localizada),[82 ] lo que es coherente con el desenlace inferior para los pacientes con DLGNT-MC-2. - Las codeleciones de 1p/19q fueron más frecuentes en el grupo de DLGNT-MC-1 (7 de 13, 54 %) que en el grupo de DLGNT-MC-2 (2 de 13, 15 %). En contraste con el oligodendroglioma, no se identificaron variantes de IDH1 y IDH2.[
80 ] - La activación de la vía MAPK es común en los casos de DLGNT.[
80 ] Se encontró la fusión KIAA1549::BRAF en 11 de 15 casos de DLGNT-MC-1 (65 %) y en 9 de 13 casos de DLGNT-MC-2 (69 %). En dos casos se encontraron fusiones que afectaron NTRK1, NTRK2 o NTRK3, y en otro caso se encontró una fusión TRIM33::RAF1.
Neurocitoma extraventricular
El neurocitoma extraventricular es similar, desde el punto de vista histológico, al neurocitoma central y contiene células pequeñas uniformes con diferenciación neuronal. Sin embargo, el neurocitoma extraventricular se presenta en el parénquima encefálico, en lugar del sistema ventricular.[
En un estudio de 40 tumores con clasificación histológica de neurocitoma extraventricular sometidos a análisis de matriz de metilación, solo 26 se agruparon según el tipo histológico en un grupo diferenciado al de los tumores de referencia de otros tipos histológicos.[
Pronóstico
Gliomas astrocíticos circunscritos, gliomas difusos de grado bajo de tipo pediátrico y tumores glioneuronales o neuronales
Por lo general, estos tumores tienen un pronóstico relativamente favorable, en particular para las lesiones bien circunscritas en las que es posible una resección radical.[
Entre las características de pronóstico clínico desfavorables se incluyen las siguientes:[
- Edad temprana.
- Incapacidad para obtener una resección completa.
- Síndrome diencefálico.
- Enfermedad diseminada o multifocal. La presencia de enfermedad diseminada o multifocal se relaciona con un desenlace a largo plazo más precario.
A nivel molecular, la presencia de una variante BRAF V600E, en especial en combinación con una deleción homocigota de CDKN2A o CDKN2B, se ha percibido como un factor pronóstico negativo, con riesgo de transformación a un tumor de grado más alto. Por el contrario, la presencia de una fusión BRAF::KIAA1549 conlleva un mejor desenlace clínico en pacientes con gliomas astrocíticos circunscritos.[
En el caso de los niños con tumores en la vía óptica, son importantes los desenlaces visuales y las evaluaciones clínicas. Los niños con tumores aislados del nervio óptico tienen un pronóstico mejor que los niños con lesiones que comprometen el quiasma o que se extienden a lo largo de la vía óptica.[
Gliomas difusos de grado alto de tipo pediátrico
Con los tratamientos disponibles en la actualidad, estos tumores tienen un pronóstico muy precario.
Los pacientes con glioma difuso de línea media con alteración H3 K27 tienen el pronóstico más precario, con tasas de supervivencia a 3 años inferiores al 5 %.[
Tumores difusos de tronco encefálico
Se usan las siguientes definiciones de tumores de tronco encefálico:
- Glioma del tronco encefálico. Término general que describe un astrocitoma que surge en el tronco encefálico. Estos tumores son circunscritos o difusos, y se presentan en cualquier lugar del tronco encefálico, incluso el mesencéfalo, la protuberancia y el bulbo raquídeo.
- Glioma pontino intrínseco difuso (GPID). Término que se usa para describir un astrocitoma infiltrante (presunto glioma difuso de línea media) centrado en la protuberancia.
- Glioma difuso de línea media con alteración H3 K27. Es el diagnóstico patológico de la mayoría de los tumores que presentan características de imágenes compatibles con un DIPG.
La mediana de supervivencia de los niños con DIPG es inferior a 1 año, aunque alrededor del 10 % sobreviven más de 2 años.[
Un informe de un ensayo clínico incluyó 42 niños y adolescentes con diagnóstico nuevo de gliomas de grado alto de línea media. En el estudio se encontró que la ubicación, el patrón de realce, la restricción de difusión y el estado de las variantes no afectó de manera significativa la supervivencia.[
Los siguientes son los factores pronósticos:
- Características histológicas o grado del tumor. Los tumores astrocíticos predominan en el tronco encefálico. Los tumores de grado 1 de la OMS (por ejemplo, astrocitomas pilocíticos y gangliogliomas) tienen un pronóstico favorable y surgen en todo el tronco encefálico, incluso en el techo del mesencéfalo, de manera focal en la protuberancia o en la unión cervicomedular, en donde son, a menudo, exofíticos. Los astrocitomas difusos de grado bajo (grado 2 de la OMS) que se presentan en otros sitios del tronco encefálico fuera de la protuberancia suelen ser tumores con un pronóstico más favorable.[
98 ]Por el contrario, los DIPG son astrocitomas difusos que, cuando se realiza una biopsia en el momento del diagnóstico, varían desde astrocitomas difusos (grado 2 de la OMS) hasta glioblastomas (grado 4 de la OMS). En la evaluación post mortem, los DIPG suelen ser astrocitomas anaplásicos (grado 3 de la OMS) o glioblastomas (grado 4 de la OMS) según criterios morfológicos, aunque también se identifican regiones correspondientes al grado 2 de la OMS.[
52 ,53 ,99 ,100 ,101 ]Cerca del 80 % de los DIPG, con independencia del grado histológico, muestran una variante de las histonas H3.3 o H3.1 y en la actualidad la OMS los clasifica como gliomas difusos de la línea media con alteración H3 K27M. Todos los gliomas difusos de la línea media con alteración H3 K27M se clasifican como de grado 4 de la OMS, con independencia del grado histológico, lo que refleja un pronóstico precario para los niños con este diagnóstico.
- Edad en el momento del diagnóstico. Se ha observado una supervivencia ligeramente más prolongada en pacientes muy jóvenes (≤3 años) o mayores (≥10 años) en el momento del diagnóstico. Cerca del 4 % de los niños con DIPG reciben el diagnóstico antes de los 3 años de edad. El pronóstico de estos niños es menos precario que el de niños mayores; el 28 % de los niños más pequeños sobreviven a los 2 años, en comparación con el 8 % de los niños de 3 a 10 años en el momento del diagnóstico y el 14 % de los niños mayores de 10 años en el momento del diagnóstico. En los niños de 10 años o más, la supervivencia a largo plazo se relacionó con una edad mayor en el momento de la presentación y con una duración de los síntomas más prolongada.[
102 ] El pronóstico más favorable para los niños pequeños tal vez refleje la presencia de características biológicas diferentes en los distintos grupos de edad.[95 ,103 ] - NF1. Es posible que los niños con NF1 y gliomas de tronco encefálico tengan un pronóstico mejor que otros pacientes con lesiones intrínsecas.[
104 ,105 ] - Características clínicas y de las imágenes en el momento del diagnóstico. En niños con DIPG, las características relacionadas con una supervivencia inferior a 2 años comprenden la presencia de parálisis de pares craneales, realce anular, necrosis y expansión por fuera de la protuberancia en el momento del diagnóstico.[
95 ] La tasa de supervivencia a 2 años es inferior al 10 % para los pacientes con estas características. - Duración de los síntomas en el momento del diagnóstico. La mayor duración de los síntomas se relaciona con un pronóstico más favorable. Las tasas de supervivencia a 2 años oscilan entre el 7 % de los pacientes con una duración de los síntomas inferior a 6 meses y el 29 % de los pacientes con una duración de los síntomas igual o superior a 24 meses.[
95 ] - Variantes de las histonas. Los pacientes con variantes H3.1 K27M tienen una mediana de supervivencia más prolongada (15 meses) que los pacientes con variantes H3.3 K27M (10,4 meses) o los pacientes sin variantes de histonas (10,5 meses).[
95 ]
Referencias:
- Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
- WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
- Kilday JP, Bartels U, Huang A, et al.: Favorable survival and metabolic outcome for children with diencephalic syndrome using a radiation-sparing approach. J Neurooncol 116 (1): 195-204, 2014.
- Klimo P, Pai Panandiker AS, Thompson CJ, et al.: Management and outcome of focal low-grade brainstem tumors in pediatric patients: the St. Jude experience. J Neurosurg Pediatr 11 (3): 274-81, 2013.
- Liu AK, Brandon J, Foreman NK, et al.: Conventional MRI at presentation does not predict clinical response to radiation therapy in children with diffuse pontine glioma. Pediatr Radiol 39 (12): 1317-20, 2009.
- Walker DA, Liu J, Kieran M, et al.: A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro Oncol 15 (4): 462-8, 2013.
- Cage TA, Samagh SP, Mueller S, et al.: Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Childs Nerv Syst 29 (8): 1313-9, 2013.
- Grill J, Puget S, Andreiuolo F, et al.: Critical oncogenic mutations in newly diagnosed pediatric diffuse intrinsic pontine glioma. Pediatr Blood Cancer 58 (4): 489-91, 2012.
- Puget S, Beccaria K, Blauwblomme T, et al.: Biopsy in a series of 130 pediatric diffuse intrinsic Pontine gliomas. Childs Nerv Syst 31 (10): 1773-80, 2015.
- Gupta N, Goumnerova LC, Manley P, et al.: Prospective feasibility and safety assessment of surgical biopsy for patients with newly diagnosed diffuse intrinsic pontine glioma. Neuro Oncol 20 (11): 1547-1555, 2018.
- Pfaff E, El Damaty A, Balasubramanian GP, et al.: Brainstem biopsy in pediatric diffuse intrinsic pontine glioma in the era of precision medicine: the INFORM study experience. Eur J Cancer 114: 27-35, 2019.
- Ryall S, Zapotocky M, Fukuoka K, et al.: Integrated Molecular and Clinical Analysis of 1,000 Pediatric Low-Grade Gliomas. Cancer Cell 37 (4): 569-583.e5, 2020.
- Buczkowicz P, Bartels U, Bouffet E, et al.: Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol 128 (4): 573-81, 2014.
- Holzapfel J, Kandels D, Schmidt R, et al.: Favorable prognosis in pediatric brainstem low-grade glioma: Report from the German SIOP-LGG 2004 cohort. Int J Cancer 146 (12): 3385-3396, 2020.
- Warren KE: Diffuse intrinsic pontine glioma: poised for progress. Front Oncol 2: 205, 2012.
- Packer RJ, Iavarone A, Jones DTW, et al.: Implications of new understandings of gliomas in children and adults with NF1: report of a consensus conference. Neuro Oncol 22 (6): 773-784, 2020.
- D'Angelo F, Ceccarelli M, Tala, et al.: The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat Med 25 (1): 176-187, 2019.
- Franz DN, Agricola K, Mays M, et al.: Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol 78 (6): 929-38, 2015.
- Mackay A, Burford A, Carvalho D, et al.: Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4): 520-537.e5, 2017.
- Bouffet E, Larouche V, Campbell BB, et al.: Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J Clin Oncol 34 (19): 2206-11, 2016.
- Das A, Tabori U, Sambira Nahum LC, et al.: Efficacy of Nivolumab in Pediatric Cancers with High Mutation Burden and Mismatch Repair Deficiency. Clin Cancer Res 29 (23): 4770-4783, 2023.
- Jones DT, Kocialkowski S, Liu L, et al.: Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68 (21): 8673-7, 2008.
- Hawkins C, Walker E, Mohamed N, et al.: BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res 17 (14): 4790-8, 2011.
- Mistry M, Zhukova N, Merico D, et al.: BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 33 (9): 1015-22, 2015.
- López GY, Van Ziffle J, Onodera C, et al.: The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol 137 (1): 139-150, 2019.
- Lassaletta A, Zapotocky M, Mistry M, et al.: Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J Clin Oncol 35 (25): 2934-2941, 2017.
- Ho CY, Mobley BC, Gordish-Dressman H, et al.: A clinicopathologic study of diencephalic pediatric low-grade gliomas with BRAF V600 mutation. Acta Neuropathol 130 (4): 575-85, 2015.
- Guerreiro Stucklin AS, Ryall S, Fukuoka K, et al.: Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun 10 (1): 4343, 2019.
- Clarke M, Mackay A, Ismer B, et al.: Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov 10 (7): 942-963, 2020.
- Meredith DM, Cooley LD, Dubuc A, et al.: ROS1 Alterations as a Potential Driver of Gliomas in Infant, Pediatric, and Adult Patients. Mod Pathol 36 (11): 100294, 2023.
- Jones DT, Hutter B, Jäger N, et al.: Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45 (8): 927-32, 2013.
- Qaddoumi I, Orisme W, Wen J, et al.: Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131 (6): 833-45, 2016.
- Zhang J, Wu G, Miller CP, et al.: Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45 (6): 602-12, 2013.
- Ramkissoon LA, Horowitz PM, Craig JM, et al.: Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 110 (20): 8188-93, 2013.
- Louis DN, Perry A, Reifenberger G, et al.: The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6): 803-20, 2016.
- Bandopadhayay P, Ramkissoon LA, Jain P, et al.: MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48 (3): 273-82, 2016.
- D'Aronco L, Rouleau C, Gayden T, et al.: Brainstem angiocentric gliomas with MYB-QKI rearrangements. Acta Neuropathol 134 (4): 667-669, 2017.
- Chan E, Bollen AW, Sirohi D, et al.: Angiocentric glioma with MYB-QKI fusion located in the brainstem, rather than cerebral cortex. Acta Neuropathol 134 (4): 671-673, 2017.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Lehman NL, Usubalieva A, Lin T, et al.: Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol Commun 7 (1): 42, 2019.
- Wood MD, Tihan T, Perry A, et al.: Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28 (2): 192-202, 2018.
- Hirose T, Nobusawa S, Sugiyama K, et al.: Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 28 (5): 684-694, 2018.
- Lucas CG, Solomon DA, Perry A: A review of recently described genetic alterations in central nervous system tumors. Hum Pathol 96: 56-66, 2020.
- Paugh BS, Qu C, Jones C, et al.: Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28 (18): 3061-8, 2010.
- Bax DA, Mackay A, Little SE, et al.: A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res 16 (13): 3368-77, 2010.
- Ward SJ, Karakoula K, Phipps KP, et al.: Cytogenetic analysis of paediatric astrocytoma using comparative genomic hybridisation and fluorescence in-situ hybridisation. J Neurooncol 98 (3): 305-18, 2010.
- Pollack IF, Hamilton RL, Sobol RW, et al.: IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children's Oncology Group. Childs Nerv Syst 27 (1): 87-94, 2011.
- Sturm D, Witt H, Hovestadt V, et al.: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 (4): 425-37, 2012.
- Korshunov A, Ryzhova M, Hovestadt V, et al.: Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol 129 (5): 669-78, 2015.
- Castel D, Kergrohen T, Tauziède-Espariat A, et al.: Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol 139 (6): 1109-1113, 2020.
- Rodriguez Gutierrez D, Jones C, Varlet P, et al.: Radiological Evaluation of Newly Diagnosed Non-Brainstem Pediatric High-Grade Glioma in the HERBY Phase II Trial. Clin Cancer Res 26 (8): 1856-1865, 2020.
- Buczkowicz P, Hoeman C, Rakopoulos P, et al.: Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet 46 (5): 451-6, 2014.
- Taylor KR, Mackay A, Truffaux N, et al.: Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46 (5): 457-61, 2014.
- Auffret L, Ajlil Y, Tauziède-Espariat A, et al.: A new subtype of diffuse midline glioma, H3 K27 and BRAF/FGFR1 co-altered: a clinico-radiological and histomolecular characterisation. Acta Neuropathol 147 (1): 2, 2023.
- Williams EA, Brastianos PK, Wakimoto H, et al.: A comprehensive genomic study of 390 H3F3A-mutant pediatric and adult diffuse high-grade gliomas, CNS WHO grade 4. Acta Neuropathol 146 (3): 515-525, 2023.
- Jain SU, Do TJ, Lund PJ, et al.: PFA ependymoma-associated protein EZHIP inhibits PRC2 activity through a H3 K27M-like mechanism. Nat Commun 10 (1): 2146, 2019.
- Hübner JM, Müller T, Papageorgiou DN, et al.: EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro Oncol 21 (7): 878-889, 2019.
- Chen CCL, Deshmukh S, Jessa S, et al.: Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 183 (6): 1617-1633.e22, 2020.
- Mackay A, Burford A, Molinari V, et al.: Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 33 (5): 829-842.e5, 2018.
- Yeo KK, Alexandrescu S, Cotter JA, et al.: Multi-institutional study of the frequency, genomic landscape, and outcome of IDH-mutant glioma in pediatrics. Neuro Oncol 25 (1): 199-210, 2023.
- Suwala AK, Stichel D, Schrimpf D, et al.: Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 141 (1): 85-100, 2021.
- Reinhardt A, Stichel D, Schrimpf D, et al.: Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 136 (2): 273-291, 2018.
- Korshunov A, Schrimpf D, Ryzhova M, et al.: H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 134 (3): 507-516, 2017.
- Becker AJ: Ganglioglioma. In: Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016, pp 138-41.
- Blumcke I, Spreafico R, Haaker G, et al.: Histopathological Findings in Brain Tissue Obtained during Epilepsy Surgery. N Engl J Med 377 (17): 1648-1656, 2017.
- Pekmezci M, Villanueva-Meyer JE, Goode B, et al.: The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6 (1): 47, 2018.
- Bianchi F, Tamburrini G, Massimi L, et al.: Supratentorial tumors typical of the infantile age: desmoplastic infantile ganglioglioma (DIG) and astrocytoma (DIA). A review. Childs Nerv Syst 32 (10): 1833-8, 2016.
- Trehan G, Bruge H, Vinchon M, et al.: MR imaging in the diagnosis of desmoplastic infantile tumor: retrospective study of six cases. AJNR Am J Neuroradiol 25 (6): 1028-33, 2004 Jun-Jul.
- Wang AC, Jones DTW, Abecassis IJ, et al.: Desmoplastic Infantile Ganglioglioma/Astrocytoma (DIG/DIA) Are Distinct Entities with Frequent BRAFV600 Mutations. Mol Cancer Res 16 (10): 1491-1498, 2018.
- Blessing MM, Blackburn PR, Krishnan C, et al.: Desmoplastic Infantile Ganglioglioma: A MAPK Pathway-Driven and Microglia/Macrophage-Rich Neuroepithelial Tumor. J Neuropathol Exp Neurol 78 (11): 1011-1021, 2019.
- Greer A, Foreman NK, Donson A, et al.: Desmoplastic infantile astrocytoma/ganglioglioma with rare BRAF V600D mutation. Pediatr Blood Cancer 64 (6): , 2017.
- Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
- Stone TJ, Keeley A, Virasami A, et al.: Comprehensive molecular characterisation of epilepsy-associated glioneuronal tumours. Acta Neuropathol 135 (1): 115-129, 2018.
- Rivera B, Gayden T, Carrot-Zhang J, et al.: Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol 131 (6): 847-63, 2016.
- Matsumura N, Nobusawa S, Ito J, et al.: Multiplex ligation-dependent probe amplification analysis is useful for detecting a copy number gain of the FGFR1 tyrosine kinase domain in dysembryoplastic neuroepithelial tumors. J Neurooncol 143 (1): 27-33, 2019.
- Pages M, Lacroix L, Tauziede-Espariat A, et al.: Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3: 85, 2015.
- Bridge JA, Liu XQ, Sumegi J, et al.: Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23 (2): 121-8, 2013.
- Hou Y, Pinheiro J, Sahm F, et al.: Papillary glioneuronal tumor (PGNT) exhibits a characteristic methylation profile and fusions involving PRKCA. Acta Neuropathol 137 (5): 837-846, 2019.
- Sievers P, Appay R, Schrimpf D, et al.: Rosette-forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co-mutation of PIK3CA and NF1. Acta Neuropathol 138 (3): 497-504, 2019.
- Deng MY, Sill M, Chiang J, et al.: Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 136 (2): 239-253, 2018.
- Chiang JCH, Harreld JH, Orr BA, et al.: Low-grade spinal glioneuronal tumors with BRAF gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol 134 (1): 159-162, 2017.
- Chiang J, Dalton J, Upadhyaya SA, et al.: Chromosome arm 1q gain is an adverse prognostic factor in localized and diffuse leptomeningeal glioneuronal tumors with BRAF gene fusion and 1p deletion. Acta Neuropathol 137 (1): 179-181, 2019.
- Sievers P, Stichel D, Schrimpf D, et al.: FGFR1:TACC1 fusion is a frequent event in molecularly defined extraventricular neurocytoma. Acta Neuropathol 136 (2): 293-302, 2018.
- Wisoff JH, Sanford RA, Heier LA, et al.: Primary neurosurgery for pediatric low-grade gliomas: a prospective multi-institutional study from the Children's Oncology Group. Neurosurgery 68 (6): 1548-54; discussion 1554-5, 2011.
- Bandopadhayay P, Bergthold G, London WB, et al.: Long-term outcome of 4,040 children diagnosed with pediatric low-grade gliomas: an analysis of the Surveillance Epidemiology and End Results (SEER) database. Pediatr Blood Cancer 61 (7): 1173-9, 2014.
- Lu VM, Di L, Gernsback J, et al.: Contemporary outcomes of diffuse leptomeningeal glioneuronal tumor in pediatric patients: A case series and literature review. Clin Neurol Neurosurg 218: 107265, 2022.
- Stokland T, Liu JF, Ironside JW, et al.: A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol 12 (12): 1257-68, 2010.
- Gnekow AK, Walker DA, Kandels D, et al.: A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (≤16 years) low grade glioma - A final report. Eur J Cancer 81: 206-225, 2017.
- Chamdine O, Broniscer A, Wu S, et al.: Metastatic Low-Grade Gliomas in Children: 20 Years' Experience at St. Jude Children's Research Hospital. Pediatr Blood Cancer 63 (1): 62-70, 2016.
- Due-Tønnessen BJ, Helseth E, Scheie D, et al.: Long-term outcome after resection of benign cerebellar astrocytomas in children and young adults (0-19 years): report of 110 consecutive cases. Pediatr Neurosurg 37 (2): 71-80, 2002.
- Massimi L, Tufo T, Di Rocco C: Management of optic-hypothalamic gliomas in children: still a challenging problem. Expert Rev Anticancer Ther 7 (11): 1591-610, 2007.
- Campagna M, Opocher E, Viscardi E, et al.: Optic pathway glioma: long-term visual outcome in children without neurofibromatosis type-1. Pediatr Blood Cancer 55 (6): 1083-8, 2010.
- Hernáiz Driever P, von Hornstein S, Pietsch T, et al.: Natural history and management of low-grade glioma in NF-1 children. J Neurooncol 100 (2): 199-207, 2010.
- Falzon K, Drimtzias E, Picton S, et al.: Visual outcomes after chemotherapy for optic pathway glioma in children with and without neurofibromatosis type 1: results of the International Society of Paediatric Oncology (SIOP) Low-Grade Glioma 2004 trial UK cohort. Br J Ophthalmol 102 (10): 1367-1371, 2018.
- Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, et al.: Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol 36 (19): 1963-1972, 2018.
- Cohen KJ, Pollack IF, Zhou T, et al.: Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro Oncol 13 (3): 317-23, 2011.
- Rodriguez D, Calmon R, Aliaga ES, et al.: MRI and Molecular Characterization of Pediatric High-Grade Midline Thalamic Gliomas: The HERBY Phase II Trial. Radiology 304 (1): 174-182, 2022.
- McAbee JH, Modica J, Thompson CJ, et al.: Cervicomedullary tumors in children. J Neurosurg Pediatr 16 (4): 357-66, 2015.
- Ballester LY, Wang Z, Shandilya S, et al.: Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol 37 (9): 1357-64, 2013.
- Wu G, Diaz AK, Paugh BS, et al.: The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46 (5): 444-50, 2014.
- Hoffman LM, DeWire M, Ryall S, et al.: Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 4: 1, 2016.
- Erker C, Lane A, Chaney B, et al.: Characteristics of patients ≥10 years of age with diffuse intrinsic pontine glioma: a report from the International DIPG/DMG Registry. Neuro Oncol 24 (1): 141-152, 2022.
- Broniscer A, Laningham FH, Sanders RP, et al.: Young age may predict a better outcome for children with diffuse pontine glioma. Cancer 113 (3): 566-72, 2008.
- Pascual-Castroviejo I, Pascual-Pascual SI, Viaño J, et al.: Posterior fossa tumors in children with neurofibromatosis type 1 (NF1). Childs Nerv Syst 26 (11): 1599-603, 2010.
- Albers AC, Gutmann DH: Gliomas in patients with neurofibromatosis type 1. Expert Rev Neurother 9 (4): 535-9, 2009.
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.