Tratamiento del neuroblastoma (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre el neuroblastoma
Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer.[
Los niños y adolescentes sobrevivientes de cáncer necesitan un seguimiento minucioso, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan, o se presenten, meses o años después de este. Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los niños y adolescentes sobrevivientes de cáncer, consultar
Incidencia y epidemiología
El neuroblastoma es el tumor sólido extracraneal más común en la infancia. Cada año se diagnostican más de 650 casos en los Estados Unidos.[
En los estudios poblacionales sobre exámenes de detección de neuroblastoma en lactantes, se observó que, durante el primer año de vida, la prevalencia de neuroblastoma sin detección clínica y con remisión espontánea es casi igual a la prevalencia del neuroblastoma que se identifica mediante evaluación clínica.[
Se usaron la base de datos de United States Cancer Statistics, así como la base de datos de supervivencia de los National Program of Cancer Registries, para describir las tendencias epidemiológicas en la incidencia y los desenlaces en pacientes con neuroblastoma de 2003 a 2019. Los pacientes blancos no hispanos tienen un mayor riesgo de presentar neuroblastoma que todos los demás grupos raciales y étnicos. En comparación con los pacientes blancos no hispanos, los riesgos relativos fueron de 0,54 para los pacientes hispanos, 0,64 para los pacientes asiáticos o de las islas del Pacífico no hispanos, 0,69 para los pacientes indígenas americanos y nativos de Alaska no hispanos y 0,73 para los pacientes negros no hispanos.[
En estudios epidemiológicos se observó que no hay exposiciones ambientales, o de otro tipo, que se relacionen de forma inequívoca con el aumento o la disminución de la incidencia del neuroblastoma.[
Características anatómicas
El neuroblastoma se origina en la médula suprarrenal, así como en las regiones paravertebrales o periaórticas donde hay tejido del sistema nervioso simpático (consultar la
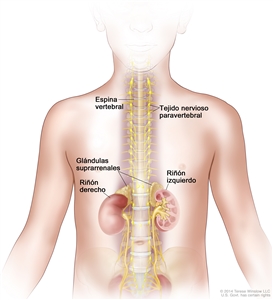
Exámenes de detección del neuroblastoma
Neuroblastoma familiar y predisposición genética
Los estudios de análisis del DNA constitucional en cohortes poco frecuentes de pacientes con neuroblastoma familiar han proporcionado información sobre las complejas bases genéticas del comienzo del tumor. Alrededor del 1 % al 2 % de los pacientes con neuroblastoma tienen antecedentes familiares de esta enfermedad. Estos niños tienen, en promedio, menor edad (9 meses en el momento del diagnóstico) que los pacientes sin antecedentes familiares y alrededor del 20 % presentan neuroblastomas primarios multifocales.
Variantes de la línea germinal. La predisposición genética al neuroblastoma se ha relacionado con varias variantes de la línea germinal, como las siguientes:
- Variante del gen ALK. La causa principal del neuroblastoma familiar (alrededor del 75 % de casos familiares) es la activación anómala de la vía de señalización germinal de ALK debido a variantes puntuales en el dominio de tirosina–cinasa del gen ALK.[
1616 ] También se observan variantes puntuales activadoras somáticas de ALK en cerca del 9 % de los casos de neuroblastoma esporádico. Además, en una proporción pequeña de casos de neuroblastoma con amplificación de MYCN, se coamplifica ALK (ALK está cerca de MYCN en el cromosoma 2), lo que también puede activar el gen ALK. ALK es un receptor tirosina–cinasa. Para más información sobre las variantes de ALK, consultar la secciónCaracterísticas genómicas y biológicas del neuroblastomaCaracterísticas genómicas y biológicas del neuroblastoma . - Variante del gen PHOX2B. En muy pocas ocasiones, el neuroblastoma familiar se relaciona con un síndrome de hipoventilación central congénita (síndrome de Ondina) que obedece a una variante de la línea germinal del gen PHOX2B.[
1717 ] La mayoría de las variantes de PHOX2B que causan el síndrome de Ondina o la enfermedad de Hirschsprung son repeticiones de polialanina que no se relacionan con el neuroblastoma familiar. Sin embargo, se identificaron variantes de la línea germinal con pérdida de función de PHOX2B en muy pocos pacientes de neuroblastoma esporádico y síndrome de Ondina o enfermedad de Hirschsprung.[1818 ] No se ha observado esta anomalía en pacientes con neuroblastoma esporádico sin síndrome de Ondina ni enfermedad de Hirschsprung relacionados. Por otra parte, las variantes somáticas de PHOX2B ocurren en cerca del 2 % de los casos de neuroblastoma esporádico.[1919 ,2020 ] - Deleción en el locus 1p36 o 11q14-23. En estudios de casos, la deleción germinal en el locus 1p36 o 11q14-23 se ha relacionado con el neuroblastoma familiar y las mismas deleciones somáticas se encuentran en algunos casos de neuroblastoma esporádico.[
2121 ,2222 ]
Otros síndromes de predisposición al cáncer. Es posible que los niños con anomalías en los genes relacionadas con otros síndromes de predisposición al cáncer tengan un riesgo más alto de presentar neuroblastoma y otras neoplasias malignas. Los síndromes que se enumeran a continuación presentan, en su gran mayoría, alteraciones en genes de la vía RAS natural:
- Síndrome de Costello.[
2323 ] - Síndrome de Noonan.[
2424 ] - Neurofibromatosis de tipo 1.[
2525 ]
Por otra parte, se ha encontrado neuroblastoma en pacientes con los siguientes síndromes:
- Síndrome de Li-Fraumeni.
- Síndromes hereditarios de feocromocitoma o paraganglioma.[
2626 ] - Síndrome de ROHHAD (obesidad de rápida progresión, disfunción hipotalámica, hipoventilación y disfunción neurovegetativa).[
2727 ] - Síndrome de Beckwith-Wiedemann.[
2828 ]
Gracias a la mayor disponibilidad de técnicas de secuenciación, se está ampliando el espectro de alteraciones de la línea germinal observadas en pacientes con neuroblastoma. Por ejemplo, en un estudio se identificó una serie de 11 pacientes con variantes patogénicas de la línea germinal en SMARCA4.[
Cabe la posibilidad de que el neuroblastoma esporádico tenga un aumento en la incidencia debido a predisposiciones germinales menos potentes. En estudios de asociación de genoma completo, se identificaron diversas variables genómicas comunes (polimorfismos mononucleótidos) que se relacionan con un riesgo más alto de presentar neuroblastoma. Gran parte de estas variables genómicas de riesgo tienen una relación significativa con fenotipos de neuroblastoma diferenciados (es decir, enfermedad de riesgo alto vs. riesgo bajo).[
Predisposición y vigilancia del neuroblastoma
Las recomendaciones para los exámenes de detección de la American Association for Cancer Research (AACR) surgieron a partir del Childhood Cancer Predisposition Workshop de 2016. La AACR recomienda que las siguientes personas se sometan a vigilancia bioquímica y radiográfica para la detección temprana de tumores durante los primeros 10 años de vida:[
- Personas con variantes hereditarias y de penetrancia alta de ALK o PHOX2B (45–50 % de riesgo de presentar uno o más tumores).
- Personas con síndrome de Li-Fraumeni y variantes germinales de TP53 p.R337H.
- Personas con síndrome de Beckwith-Wiedemann y variantes germinales de CDKN1C.
- Personas con síndrome de Costello y variantes de HRAS.
- Personas con neuroblastoma y antecedentes familiares marcados de neuroblastoma, o neuroblastoma bilateral o multifocal evidente.
La vigilancia se realiza mediante los siguientes procedimientos:[
- Ecografía abdominal.
- Evaluación cuantitativa y normalizada de catecolaminas urinarias,[
3232 ] como el ácido vanililmandélico (VMA) y el ácido homovanílico (HVA) urinarios, mediante cromatografía de gases y espectroscopia de masas (por ejemplo, análisis de muestra de orina al azar normalizada según la creatinina urinaria porque este abordaje parece que tiene una sensibilidad similar a la recolección de 24 horas). - Radiografía del tórax.
La vigilancia comienza en el momento del nacimiento o del diagnóstico de la predisposición al neuroblastoma y continúa cada 3 meses hasta los 6 años de edad, y luego cada 6 meses hasta los 10 años de edad. En ocasiones, los pacientes con síndrome de Costello tienen una concentración urinaria alta de catecolaminas en ausencia de un tumor que segrega catecolaminas; por lo tanto, solo cuando esta concentración sea muy alta o aumente mucho de manera progresiva se contemplarán otros estudios, además de la ecografía y la radiografía del tórax.[
Alrededor del 5 % de los niños con síndrome de Beckwith-Wiedemann presentan variantes que producen una disminución de la actividad de CDKN1C. En una revisión de todos los estudios grandes sobre subtipos genéticos de síndrome de Beckwith-Wiedemann se hallaron 70 niños con variante de CDKN1C, de los cuales el 4,6 % presentaron neuroblastoma. No hubo casos de tumor de Wilms ni de hepatoblastoma. Hay poca experiencia respecto a los exámenes de detección de neuroblastoma en estos niños, por lo que no existen pautas de aceptación general. No obstante, los autores del estudio recomiendan que se realicen exámenes de detección con VMA/HVA urinario cada 4 o 6 meses. Los pacientes con otros subtipos genéticos de síndrome de Beckwith-Wiedemann tienen una prevalencia de neuroblastoma inferior al 1 %. No se encontraron tumores neuroblásticos entre los 123 niños con el genotipo de ganancia de metilación en la región 1 de control de impronta.[
Población general
Los datos actualizados no respaldan el uso de exámenes de detección del neuroblastoma para la población general. Los exámenes de detección a las 3 semanas, 6 meses o 1 año de vida no conducen a una reducción de la incidencia de neuroblastoma en estadio avanzado con características biológicas desfavorables en niños mayores, ni disminuye la mortalidad general por neuroblastoma.[
Evidencia (en contra de los exámenes de detección del neuroblastoma):
- En un estudio poblacional grande en América del Norte, la mayoría de los lactantes de Quebec (Canadá) se sometieron a un examen de detección a las 3 semanas y a los 6 meses de edad.[
1212 ,1313 ]- En el examen de detección se observaron muchos neuroblastomas con características favorables que nunca se habrían detectado mediante evaluación clínica debido a la remisión espontánea de los tumores.
- En otro estudio de lactantes sometidos a exámenes de detección al año de edad se observaron resultados similares.[
1414 ]
Cuadro clínico inicial
Los signos y síntomas más frecuentes del neuroblastoma en niños se deben a la masa tumoral y las metástasis, y comprenden los que siguen a continuación.
- Masa abdominal: este es el cuadro clínico inicial más común del neuroblastoma.
- Proptosis y equimosis periorbitaria: comunes en pacientes de riesgo alto; surgen de una metástasis retrobulbar.
- Distensión abdominal: en lactantes se puede presentar con compromiso respiratorio debido a metástasis hepáticas masivas.
- Dolor óseo: se presenta vinculado con la enfermedad metastásica.
- Pancitopenia: puede ser consecuencia de metástasis extensas en la médula ósea.
- Fiebre, hipertensión y anemia: en ocasiones, se encuentran en pacientes sin metástasis.
- Parálisis: el neuroblastoma que se origina en los ganglios paravertebrales a veces invade a través de los agujeros intervertebrales y causa compresión extradural de la médula espinal. Se administra tratamiento inmediato para la compresión sintomática de la médula espinal. Para obtener más información, consultar la sección
Tratamiento de la compresión de la médula espinalTratamiento de la compresión de la médula espinal . - Diarrea acuosa: con escasa frecuencia, los niños pueden presentar diarrea acuosa grave porque el tumor segrega péptido intestinal vasoactivo o por enteropatía con pérdida de proteínas y linfangiectasia intestinal.[
3535 ] Es posible que la segregación de péptido intestinal vasoactivo ocurra en el momento de la presentación (siendo la diarrea el primer síntoma de neuroblastoma), al inicio de la quimioterapia, o más adelante, durante el curso del tratamiento. La resección del tumor reduce la segregación de péptido intestinal vasoactivo.[3636 ] - Presencia de síndrome de Horner: se caracteriza por miosis, ptosis y anhidrosis. Esto puede obedecer a un neuroblastoma en el ganglio estrellado; los niños con síndrome de Horner sin otra causa aparente también se examinan para identificar el neuroblastoma y otros tumores.[
3737 ] - Nódulos subcutáneos: las metástasis subcutáneas del neuroblastoma a menudo tienen coloración azulada en la piel suprayacente; estos nódulos se suelen observar solo en lactantes.
El cuadro clínico inicial del neuroblastoma en adolescentes es similar al de los niños. La única excepción es que el compromiso de médula ósea se presenta con menos frecuencia en los adolescentes y que la metástasis es más frecuente en sitios poco habituales, como el pulmón o el encéfalo.[
Síndrome opsoclono-mioclono
En casos infrecuentes, los niños con neuroblastoma presentan manifestaciones neurológicas paraneoplásicas, como ataxia cerebelosa, opsoclonía o mioclonía.[
El cuadro clínico por lo general incluye aparición de disfunción neurológica progresiva durante unos pocos días antes del hallazgo de un neuroblastoma; sin embargo, a veces, los síntomas neurológicos surgen mucho después de la extirpación del tumor primario.[
El síndrome opsoclono-mioclono parece obedecer a un mecanismo inmunitario que todavía no está bien definido.[
Se analizaron los perfiles del número de copias genómicas en 44 casos de neuroblastoma relacionados con el síndrome opsoclono-mioclono. Dado que no se produjeron recaídas tumorales ni muertes relacionadas con la enfermedad, el perfil genómico global no fue significativo desde el punto de vista pronóstico.[
Algunos pacientes presentan una respuesta neurológica rápida a intervenciones inmunitarias o, simplemente, a la extirpación del neuroblastoma, pero en muchos casos la mejora es lenta y parcial. La mejora observada en las manifestaciones iniciales de déficit motor y ataxia en respuesta a la terapia inmunológica, no se relaciona de manera clara con la mejora de la discapacidad neuropsicológica a largo plazo, que consiste sobre todo en un deterioro cognitivo y conductual. No están claros los beneficios a largo plazo de la mejora rápida con el tratamiento, ya sea en los síntomas o en el neuroblastoma subyacente, pero este tratamiento vale la pena debido a la rápida mejora observada.[
El tratamiento con hormona adrenocorticotrópica o corticoesteroides a veces tiene efecto en los síntomas agudos, pero algunos pacientes no responden a los corticoesteroides.[
El Children's Oncology Group (COG) llevó a cabo el primer estudio aleatorizado y sin anonimato de fase III de pacientes con síndrome opsoclono-mioclono-ataxia.[
- De los 53 pacientes que participaron, 21 de 26 (81 %) del grupo de IVIG presentaron respecto del síndrome opsoclono-mioclono-ataxia durante un período de semanas a meses, en comparación con 11 de 27 pacientes (41 %) en el grupo sin IVIG (oportunidad relativa [OR], 6,1; P = 0,0029).
- En este estudio se demostró que la respuesta neurológica a corto plazo mejora en pacientes tratados con quimioterapia, corticoesteroides e inmunoglobulina, en comparación con pacientes tratados con quimioterapia y corticoesteroides sin inmunoglobulina.
- Se requiere de seguimiento adicional para evaluar los problemas a largo plazo de desarrollo neurológico y aprendizaje en esta población.
Diagnóstico
La evaluación diagnóstica del neuroblastoma incluye los siguientes procedimientos.
- Imágenes del tumor: por lo general, las imágenes de la masa tumoral primaria se obtienen mediante tomografía computarizada o imágenes por resonancia magnética (IRM) con contraste. Los tumores paraespinales que están a punto de comprimir la médula espinal se estudian mediante IRM.
La gammagrafía con metayodobencilguanidina (MIBG) es una parte esencial de la evaluación diagnóstica estándar del neuroblastoma, tanto para el tumor primario como para los sitios de metástasis.[
5656 ,5757 ] Asimismo, la gammagrafía con MIBG es esencial para evaluar la respuesta al tratamiento.[5757 ] Cerca del 90 % de los casos de neuroblastoma son ávidos de MIBG; Se usan tomografías por emisión de positrones (TEP) con flúor F 18-fluorodesoxiglucosa para evaluar la extensión de la enfermedad en pacientes con tumores que no sean ávidos de MIBG.[5858 ] Para obtener más información sobre pruebas con imágenes del neuroblastoma, consultar la secciónInformación sobre los estadios del neuroblastomaInformación sobre los estadios del neuroblastoma . - Metabolitos de las catecolaminas en la orina: antes del tratamiento, se mide la excreción urinaria de los metabolitos de las catecolaminas VMA y HVA por miligramo de creatinina excretada. No es necesaria la recolección de orina durante 24 horas. Si estos marcadores permanecen elevados, indican persistencia de la enfermedad.
Las concentraciones séricas de catecolaminas, en contraste con las concentraciones en orina, no se utilizan de rutina para el diagnóstico del neuroblastoma , excepto en circunstancias no habituales.
- Biopsia: en los ensayos clínicos actuales del COG, con frecuencia se necesita extraer tejido tumoral para obtener todos los datos biológicos necesarios con el fin de asignar el grupo de riego y determinar la estratificación posterior del tratamiento. La obtención de una biopsia tisular es un requisito definitivo para determinar la
International Neuroblastoma Pathology ClassificationInternational Neuroblastoma Pathology Classification (INPC). Además, se necesita una cantidad importante de células tumorales para determinar el número de copias de MYCN, el índice de DNA y la presencia de anomalías cromosómicas segmentarias. De igual forma, es necesario tejido de varias biopsias con aguja, o aproximadamente 1 cm3 de tejido de una biopsia abierta para una estadificación biológica correcta.En los pacientes mayores de 18 meses con enfermedad en estadio 4, el compromiso tumoral extenso de la médula ósea combinado con metabolitos de catecolaminas elevados quizás resulte adecuado para el diagnóstico y la asignación del grupo de riesgo o tratamiento. Sin embargo, la INPC no se puede determinar a partir de un tumor metastásico en la médula ósea. Es posible realizar con éxito una prueba de amplificación de MYCN en la médula ósea comprometida si el compromiso tumoral alcanza, al menos, el 30 %. No obstante, debe hacerse todo lo posible por obtener una biopsia adecuada del tumor primario.
Para obtener información sobre el uso de biopsias en pacientes menores de 1 año de edad, consultar la sección
Observación y remisión espontánea del neuroblastoma fetal o neonatalObservación y remisión espontánea del neuroblastoma fetal o neonatal .
El diagnóstico del neuroblastoma exige la participación de patólogos familiarizados con los tumores infantiles. Algunos neuroblastomas no se diferencian morfológicamente solo mediante microscopía óptica convencional y tinción con hematoxilina y eosina de otros tumores de células pequeñas, redondas y azules de la niñez, como los linfomas, el sarcoma de Ewing y los rabdomiosarcomas. En estos casos, tal vez sean necesarios análisis inmunohistoquímicos y citogenéticos para diagnosticar un tumor específico de células azules, redondas y pequeñas.
El criterio mínimo, establecido por acuerdo internacional, para diagnosticar el neuroblastoma se basa en una de las características siguientes:
- Un diagnóstico patológico inequívoco mediante análisis de tejido tumoral con microscopia óptica (con estudio inmunohistológico o microscopía electrónica, o sin estas).[
5959 ] - La identificación inequívoca de células tumorales (por ejemplo, sincitios o racimos de células inmunocitológicamente positivas) en un aspirado de médula ósea combinado con biopsia por trépano y concentraciones elevadas de metabolitos de catecolaminas en la orina.[
5959 ]
Observación y remisión espontánea del neuroblastoma fetal o neonatal
El fenómeno de remisión espontánea se ha descrito bien en lactantes con neuroblastoma, en particular en quienes presentan el patrón de diseminación metastásica del estadio 4S del INSS o MS del INRG.[
Por lo general, la remisión espontánea se presenta en tumores con las siguientes características:[
- Número de cromosomas casi triploide.
- Ausencia de amplificación de MYCN.
- No hay pérdida del cromosoma 1p.
Otras características relacionadas con una remisión espontánea incluyen la ausencia de expresión de la telomerasa,[
En ciertos estudios se ha indicado que algunos lactantes asintomáticos que tienen un neuroblastoma suprarrenal pequeño de grado bajo, identificado mediante un examen de detección o de forma casual durante una ecografía prenatal, a menudo presentan tumores con regresión espontánea y se pueden vigilar de manera inocua sin intervención quirúrgica o diagnóstico tisular.[
Evidencia (observación [remisión espontánea]):
- En un estudio del COG, se hizo un seguimiento con observación sin biopsia de 83 lactantes menores de 6 meses seleccionados que tenían masas suprarrenales pequeñas en estadio 1 (3,1 cm o menos) conforme a estudios de imágenes. La intervención quirúrgica se reservó para aquellos que presentaban crecimiento o progresión de la masa, o aumento de las concentraciones de metabolitos de catecolaminas en la orina.[
7171 ]- No se sometió a cirugía al 81 % de los pacientes y todos ellos estaban vivos tras 2 años de seguimiento. Para obtener más información, consultar la sección
CirugíaCirugía . - En consecuencia, es posible hacer un seguimiento inocuo de las masas suprarrenales que se detectan en el periodo prenatal y que miden como máximo 3,1 cm, si no se identifica enfermedad metastásica ni compromiso de vasos grandes o de órganos.
- No se sometió a cirugía al 81 % de los pacientes y todos ellos estaban vivos tras 2 años de seguimiento. Para obtener más información, consultar la sección
- En un ensayo clínico alemán se informó sobre 340 lactantes con neuroblastoma localizado sin amplificación de MYCN. De estos pacientes, 190 se sometieron a resección, 57 recibieron quimioterapia y en 93 se detectó tumor residual macroscópico.[
7272 ]- Se presentó remisión espontánea o ausencia de progresión tumoral en 44 de 93 lactantes asintomáticos, de 12 meses o menos, con tumores en estadios 1, 2 o 3 sin amplificación de MYCN.
- Todos los pacientes se sometieron a observación después de una biopsia y una resección parcial o sin esta.
- En algunos casos, la remisión ocurrió más de un año después del diagnóstico.
- En ensayos de exámenes de detección del neuroblastoma realizados en Quebec (Canadá) y en Alemania, la incidencia del neuroblastoma fue el doble de la notificada sin exámenes de detección, lo cual indica que muchos neuroblastomas nunca se diagnostican mediante evaluación clínica y son de regresión espontánea.[
1212 ,1313 ,1414 ]
Factores pronósticos
El pronóstico de los pacientes con neuroblastoma se relaciona con los siguientes aspectos:
-
Período de tratamientoPeríodo de tratamiento . -
Edad en el momento del diagnósticoEdad en el momento del diagnóstico . -
Características histológicas del tumorCaracterísticas histológicas del tumor . -
Características biológicasCaracterísticas biológicas . -
Sitio del tumor primarioSitio del tumor primario . -
Estadio de la enfermedadEstadio de la enfermedad . -
Respuesta al tratamientoRespuesta al tratamiento . -
Concentraciones de lactato–deshidrogenasa y ferritinaConcentraciones de lactato–deshidrogenasa y ferritina .
Se combinaron algunos de estos factores pronósticos para crear grupos de riesgo a fin de definir el tratamiento. Para obtener más información, consultar las secciones
Período de tratamiento
En los Estados Unidos, entre 1975 y 2017, la tasa de supervivencia a 5 años para el neuroblastoma aumentó del 86 % al 91 % en niños menores de 1 año, y del 34 % al 83 % en niños de 1 a 14 años.[
Edad en el momento del diagnóstico
Lactantes y niños
El efecto de la edad en el momento del diagnóstico en la supervivencia a 5 años es pronunciado. En el estudio del COG ANBL00B1 (NCT00904241) de 4832 pacientes con neuroblastoma recién diagnosticado, los menores de 18 meses presentaron una tasa de SSC a 5 años del 82 % y una tasa de SG del 91 %. En comparación, los pacientes de 18 meses o más tuvieron una tasa de SSC a 5 años del 64 % y una tasa de SG del 74 %.[
Según el National Childhood Cancer Registry (NCCR), las tasas relativas de supervivencia a 5 años de 2011 a 2017 fueron las siguientes:[
- Edad menor de 1 año: 91 %
- Edad de 1 y 4 años: 79 %
- Edad de 5 y 9 años: 79 %
- Edad de 10 y 14 años: 91 %
Los factores clínicos y biopatológicos influyen sobremanera en el efecto que tiene la edad del paciente sobre el pronóstico, según se muestra a continuación:
- Desde 2000, en estudios no aleatorizados de pacientes de riesgo bajo e intermedio se demostró que la edad del paciente no afecta el desenlace de la enfermedad en estadios 1 o 2A del INSS. Sin embargo, los pacientes en estadio 2B menores de 18 meses tuvieron una tasa de SG a 5 años del 99 % (± 1 %) versus el 90 % (± 4 %) de los niños de 18 meses o más.[
7777 ] - En el estudio de riesgo intermedio del COG A3961 (NCT00003093) que solo incluyó tumores sin amplificación de MYCN, los lactantes con tumores en estadio 3 del INSS se compararon con niños con tumores de tipo histológico favorable en estadio 3 del INSS. Cuando se hizo una comparación entre los lactantes con tumores en estadio 3 del INSS de cualquier tipo histológico y los niños con tumores en estadio 3 de tipo histológico favorable, solo las tasas de SSC, pero no las tasas de SG, fueron diferentes en forma significativa (tasa de SSC a 3 años, 95 % ± 2 vs. 87 % ± 3 %; tasa de SG, 98 % ± 1 vs. 99 % ± 1 %).[
7878 ] - Los lactantes menores de 12 meses con enfermedad en estadio 4 del INSS y amplificación de MYCN se clasifican como de riesgo alto y tienen una tasa de SSC a 5 años del 37 % y una tasa de SG del 45 %.[
7676 ] Los niños de 12 meses a 18 meses con enfermedad en estadio 4 y tumores con amplificación de MYCN tuvieron una tasa de SSC a 5 años del 53 % y una tasa de SG del 54 %.[7676 ]
Adolescentes y adultos jóvenes
El neuroblastoma es infrecuente en adolescentes y adultos, quienes representan menos del 5 % de todos los casos. Cuando se presenta un neuroblastoma en este grupo etario, tiene una evolución clínica poco activa (de crecimiento lento, escasa malignidad, indolente) que el neuroblastoma en los pacientes más jóvenes, y exhibe resistencia de novo a la quimioterapia.[
Aunque la amplificación de MYCN es infrecuente en los pacientes adolescentes y adultos jóvenes (un 9 % en pacientes de 10 a 21 años de edad), los niños mayores con enfermedad avanzada tienen una tasa de supervivencia precaria. Es habitual que los tumores en la población de adolescentes y adultos jóvenes exhiban anomalías cromosómicas segmentarias, y que las variantes de ALK y ATRX sean mucho más frecuentes.[
La tasa de SG a 5 años en pacientes adolescentes y adultos jóvenes (15–39 años) es del 38 %.[
Adultos
Las características biológicas de los neuroblastomas que inician en la adultez son diferentes de las de los neuroblastomas en la niñez y adolescencia, de acuerdo con una serie de una sola institución de 44 pacientes (edad, 18–71 años).[
- Las anormalidades genéticas en los pacientes adultos incluyen variantes somáticas de ATRX (58 %) y ALK (42 %), pero no amplificaciones de MYCN.
- Se realizaron pruebas de la línea germinal en 4 pacientes, 2 de los cuales presentaron anomalías (un paciente con una variante de BRCA1 y el otro paciente con variantes de TP53 y NF1.
- En los 11 pacientes con enfermedad locorregional, la tasa de supervivencia sin progresión (SSP) a 10 años fue del 35 %, y la tasa de SG fue del 61 %.
- Entre 33 pacientes adultos con neuroblastoma en estadio 4, 7 (21 %) lograron una respuesta completa (RC) después de la quimioterapia de inducción o cirugía. En los pacientes con diagnóstico de enfermedad en estadio 4, la tasa de SSP a 5 años fue del 10 %, y la mayoría de los pacientes vivos que todavía tenían la enfermedad al cabo de 5 años murieron debido al neuroblastoma en el transcurso de los 5 años siguientes; la tasa de SG a 10 años fue del 19 %. La RC después de la inducción fue el único factor pronóstico de la SSP y SG.
- Los adultos toleraron bien la inmunoterapia anti-GD2 (m3F8 o hu3F8).
Como se indicó antes, en el neuroblastoma que se forma durante la edad adulta predominan las variantes activadoras de ALK. En un estudio retrospectivo de una sola institución, 13 pacientes adultos (mediana de edad, 34 años; intervalo, 16–71 años) con neuroblastoma en recaída, que presentaba variante de ALK, se trataron con lorlatinib. De los pacientes, 9 (69 %) tuvieron respuestas completas o parciales; 5 de ellos ya habían recibido tratamiento con inhibidores de ALK. El lorlatinib se relacionó con efectos adversos importantes que requirieron reducción de la dosis. Sin embargo, se observaron respuestas con dosis menores a las recomendadas para los adultos.[
Características histológicas del tumor
El tipo histológico del tumor del neuroblastoma tiene un efecto significativo en el pronóstico y la asignación del grupo de riesgo. Para obtener más información, consultar la sección
En el estudio ANBL00B1 (NCT00904241) de 4832 pacientes con neuroblastoma recién diagnosticado, el 52 % de los pacientes se clasificaron como favorables y el 48 % como desfavorables, según la International Neuroblastoma Pathology Classification (INPC). La tasa de SSC a 5 años de los pacientes con tumores clasificados como favorables fue del 88 % y la tasa de SG a 5 años fue del 96 %. Para los pacientes con tumores clasificados como desfavorables, la tasa de SSC a 5 años fue del 55 % y la tasa de SG a 5 años fue del 66 % (P < 0,0001).[
Las características histológicas que se consideran favorables desde el punto de vista pronóstico son las siguientes:
- Diferenciación y maduración celular. Los grados más elevados de maduración neuroblástica confieren un mejor pronóstico para los pacientes en estadio 4 que exhiben cambios cromosómicos segmentarios sin amplificación de MYCN. Los neuroblastomas que contienen muchas células diferenciadas se llaman ganglioneuroblastomas y a veces tienen una diferenciación difusa que confiere un pronóstico muy favorable. Cuando los neuroblastomas tienen nódulos de células no diferenciadas, se denominan ganglioneuroblastomas nodulares, cuyo tipo histológico, junto con el estado de MYCN, determina el pronóstico.[
8989 ,9090 ] - Estroma schwanniano.
- Neuroblastoma quístico. Alrededor del 25 % de los neuroblastomas diagnosticados en el período fetal o neonatal que se notifican son quísticos; los pacientes con este tipo de neuroblastoma presentan tumores con una estadificación más baja y una incidencia más alta de características biológicas favorables.[
9191 ]
Un índice alto de mitosis-cariorrexis y células tumorales indiferenciadas se consideran características histológicas desfavorables para el pronóstico, pero el valor pronóstico depende de la edad.[
En un estudio del COG (P9641 [NCT00003119]), en el que se investigaron los efectos del tipo histológico, entre otros factores, en el desenlace, se trató con cirugía inicial y observación al 87 % de 915 niños con neuroblastoma en estadios 1 y 2 sin amplificación de MYCN. Los pacientes (13 %) con enfermedad sintomática o en riesgo de presentarla, los pacientes con resección tumoral inferior al 50 % en el momento del diagnóstico, y los pacientes con enfermedad progresiva irresecable después de la cirugía sola, se trataron con quimioterapia y cirugía. Aquellos con características histológicas favorables notificaron una tasa de SSC a 5 años del 90 % al 94 % y una tasa de SG del 99 % al 100 %. Los que presentaron tipo histológico desfavorable tuvieron una tasa de SSC a 5 años del 80 % al 86 % y una tasa de SG del 89 % al 93 %.[
- En el estudio del COG ANBL0531 (NCT00499616) de pacientes de riesgo intermedio con neuroblastoma, el tratamiento se asignó mediante un algoritmo que tuvo en cuenta las características biológicas y las respuestas, incluso el estado alélico de 1p36 y 11q23. Se excluyeron los pacientes que presentaban tumores con amplificación de MYCN.[
7575 ]- La SSC fue significativamente mejor, desde el punto de vista estadístico, en los lactantes con enfermedad en estadio 4 y tumor de características biológicas favorables (n = 61) (tasa de SSC a 3 años, 86,9 %; intervalo de confianza [IC] 95 %, 78,3–95,4 %), en comparación con aquellos que presentaron tumor de características biológicas desfavorables confirmadas (n = 47) (tasa de SSC a 3 años, 66,8 %; IC 95 %, 53,1–80,6 %; P = 0,02).
- En la SG de los lactantes con enfermedad en estadio 4 y tumor de características biológicas favorables se observó una tendencia hacia una SG mejor (tasa de SG a 3 años,95,0 %, IC 95 %, 89,5–100 % vs. 86,7 %; IC 95 %, 76,6–96,7 %; P = 0,08).
- Entre los lactantes del grupo, 4 lactantes (n = 24) con enfermedad en estadio 4 y tumores diploides o de tipo histológico desfavorable confirmados, con pérdida de heterocigosis en 1p36/11q23 o sin esta, presentaron una tasa estimada de SSC a 3 años del 63,9 % (IC 95 %, 43,8–84,0 %) y una tasa estimada de SG a 3 años del 77,3 % (IC 95 %, 59,2–95,3 %).
- En los lactantes con tumores hiperdiploides y tipo histológico favorable en estadio 4 que se asignaron al grupo 4, debido a la pérdida de heterocigosis en 1p36/11q23 o estado alélico desconocido (n = 32), las tasas estimadas de SSC y SG a 3 años fueron del 68,6 % (IC 95 %, 52,2–85,1 %) y el 93,8 % (IC 95 %, 85,2–100 %), respectivamente.
- Las tasas estimadas de SSC y SG de 8 niños (12–18 meses de edad) con tumores hiperdiploides de tipo histológico favorable en estadio 4 fueron del 62,5 % (IC 95 %, 28,9–96,1 %) y el 100 %, respectivamente.
- Los pacientes con características biológicas favorables y enfermedad localizada tuvieron una tasa de supervivencia del 100 %.
En un estudio en el que se usaron datos del INRG Data Commons se evaluó el alcance pronóstico de los criterios histológicos subyacentes del INPC. Se demostró la capacidad pronóstica independiente de la edad, la categoría histológica, el índice de mitosis-cariorrexis (MKI) y el grado. Se identificaron 4 grupos de pronóstico histológico relacionados con la edad (edad <18 meses con MKI bajo vs. alto, y edad ≥18 meses con tumores diferenciados vs. indiferenciados o poco diferenciados). En comparación con los árboles de supervivencia generados con los criterios de riesgo del COG establecidos, se identificó y validó un subgrupo pronóstico adicional cuando se analizaron las características histológicas individuales en lugar de usar la INPC. Por lo tanto, la sustitución de la INPC por características histológicas individuales en la futura clasificación de riesgo del COG tal vez elimine la duplicación de la contribución pronóstica de la edad, facilite la armonización internacional de la clasificación de riesgo y proporcione un esquema para un pronóstico más preciso y abordajes terapéuticos refinados.[
Características biológicas
Para obtener más información, consultar la sección
Sitio del tumor primario
Las características clínicas y biológicas del neuroblastoma difieren según el sitio del tumor primario. En un estudio con datos de 8389 pacientes que participaron en ensayos clínicos y que fueron recopilados por el International Risk Group Project, se observaron los siguientes resultados que confirman los resultados de estudios anteriores mucho más pequeños, con menos datos clínicos y biológicos completos:[
- Tumores suprarrenales. Fue más probable que los tumores primarios suprarrenales, en comparación con los tumores originados en otros sitios, exhibieran características pronósticas desfavorables, como amplificación de MYCN, incluso después de que los investigadores introdujeran controles por edad, estadio y grado histológico. Los neuroblastomas suprarrenales también se relacionaron con una incidencia más alta de tumores en estadio 4, anomalías cromosómicas segmentarias, diploidía, tipo histológico de la INPC desfavorable, edad menor de 18 meses, y concentraciones elevadas de LDH y ferritina. El riesgo relativo de una amplificación de MYCN, comparado con el de tumores suprarrenales, fue de 0,7 en tumores abdominales no suprarrenales y de cerca de 0,1 en tumores paravertebrales extraabdominales.
- Tumores torácicos. Los tumores torácicos se compararon con los tumores no torácicos. Después de que los investigadores realizaran ajustes por edad, estadio y grado histológico, los resultados mostraron menos muertes y recidivas en los pacientes con tumores torácicos (CRI, 0,79; IC 95 %, 0,67–0,92) y una incidencia más baja de amplificación de MYCN (OR ajustada, 0,20; IC 95 %, 0,11–0,39) en dichos tumores.
En un estudio se usaron los Therapeutically Applicable Research to Generate Effect Treatments (TARGET) y los conjuntos de datos de estudios de asociación de genoma completo para comparar los datos genómicos y epigenómicos de neuroblastomas de diagnóstico primario que surgen en la glándula suprarrenal (n = 646) y los neuroblastomas que surgen en ganglios simpáticos torácicos (n = 118). Los neuroblastomas que surgieron en la glándula suprarrenal fueron más propensos a albergar anomalías estructurales del DNA, como la amplificación de MYCN, mientras que los tumores torácicos mostraron defectos en los puntos de control mitótico que daban lugar a hiperdiploidía. Los tumores torácicos fueron más propensos a portar anomalías de ganancia de función en ALK, en comparación con los tumores suprarrenales en todos los casos (OR, 1,89; P = 0,04), y en los casos sin amplificación de MYCN (OR, 2,86; P = 0,003). Debido a que el 16 % de los tumores torácicos portan variantes de ALK, se debe considerar la secuenciación rutinaria para dichas variantes en este contexto.[
En la cohorte TARGET, el 70 % de los pacientes con tumores primarios suprarrenales y el 51 % de los pacientes con tumores primarios torácicos presentaban enfermedad en estadio 4. En el estudio de asociación de genoma completo sin amplificación de MYCN, el 43 % de los pacientes con tumores primarios suprarrenales y el 17 % de los pacientes con tumores primarios torácicos presentaban enfermedad en estadio 4. Según el análisis multivariante, la localización suprarrenal fue un factor de predicción independiente de desenlaces más precarios en el estudio de asociación de genoma completo, pero no en la cohorte TARGET después de que se ajustara por el estado de amplificación de MYCN, estadio de la enfermedad y edad de, al menos, 18 meses. En otro análisis multivariante similar al estudio de asociación de genoma completo y de la cohorte TARGET, se observó que el neuroblastoma suprarrenal no era un factor de predicción independiente de una SSC más precaria.[
No queda claro si el efecto pronóstico del sitio del tumor primario del neuroblastoma depende completamente de las diferencias en las características biológicas del tumor según el sitio tumoral.
El neuroblastoma multifocal se presenta con poca frecuencia, a menudo en lactantes, y, por lo general, tiene buen pronóstico.[
Estadio de la enfermedad
Antes de la década de 1990, se utilizaban varios sistemas con imágenes y sistemas quirúrgicos para asignar el estadio de la enfermedad. En un esfuerzo por facilitar la comparación de los resultados obtenidos en todo el mundo, se creó un sistema de estadificación patológica y quirúrgica, conocido como el International Neuroblastoma Staging System (INSS).[
Para los pacientes con neuroblastoma recién diagnosticado inscritos en el estudio ANBL00B1 (NCT00904241), las tasas de SSC y SG a 5 años, según la estadificación del INRGSS, fueron las siguientes:[
- Del 90 % y el 98 % para pacientes con enfermedad en estadio L1.
- Del 84 % y el 95 % para pacientes con enfermedad en estadio L2.
- Del 52 % y el 64 % para pacientes con enfermedad en estadio M.
- Del 86 % y el 92 % para pacientes con enfermedad en estadio MS.
Para obtener más información, consultar las siguientes secciones:
-
International Neuroblastoma Staging SystemInternational Neuroblastoma Staging System . -
International Neuroblastoma Risk Group Staging SystemInternational Neuroblastoma Risk Group Staging System . -
Opciones de tratamiento para el neuroblastoma de riesgo bajo, evidencia (para la eliminación de la quimioterapia) Opciones de tratamiento para el neuroblastoma de riesgo bajo, evidencia (para la eliminación de la quimioterapia) . -
Opciones de tratamiento del neuroblastoma de riesgo intermedio, evidencia (quimioterapia con cirugía o sin esta)Opciones de tratamiento del neuroblastoma de riesgo intermedio, evidencia (quimioterapia con cirugía o sin esta) . -
Opciones de tratamiento del neuroblastoma de riesgo intermedio, radioterapiaOpciones de tratamiento del neuroblastoma de riesgo intermedio, radioterapia . -
Opciones de tratamiento del neuroblastoma de riesgo altoOpciones de tratamiento del neuroblastoma de riesgo alto . -
Opciones de tratamiento del neuroblastoma en estadio 4S o MSOpciones de tratamiento del neuroblastoma en estadio 4S o MS .
Respuesta al tratamiento
La respuesta al tratamiento se ha relacionado con el desenlace. En pacientes con enfermedad de riesgo intermedio que tuvieron una respuesta precaria a la terapia inicial en el estudio del COG ANBL0531 (NCT00499616), 6 de 20 pacientes presentaron enfermedad recidivante o progresiva, y 1 paciente murió.[
Por ejemplo, en los pacientes con enfermedad de riesgo alto, la persistencia de células de neuroblastoma en la médula ósea después de la administración de quimioterapia de inducción se relaciona con un pronóstico precario. El pronóstico se puede evaluar mediante técnicas sensibles a la enfermedad residual mínima.[
En un análisis de pacientes de 4 ensayos consecutivos de riesgo alto del COG, una respuesta parcial (RP) o mejor al final de la inducción, según los International Neuroblastoma Response Criteria de 1993,[
Una disminución de las mitosis y un aumento de la diferenciación histológica del tumor primario después del tratamiento también predicen la respuesta.[
La exactitud del pronóstico a partir de la disminución del tamaño del tumor primario es menos clara. En un estudio realizado en 7 centros internacionales grandes, 229 pacientes de riesgo alto recibieron diversas formas de tratamiento, como quimioterapia, extirpación quirúrgica del tumor primario, radiación dirigida al lecho tumoral, terapia mielosupresora con trasplante de células madre y, en la mayoría de los casos, isotretinoína e inmunoterapia con un anticuerpo anti-GD2 potenciada con citocinas. La respuesta del tumor primario después de la quimioterapia de inducción se midió de 3 maneras: reducción del 30 % o más en la dimensión más larga del tumor, reducción del 50 % o más del volumen tumoral, o una reducción del 65 % o más del volumen tumoral (calculada con una técnica radiológica convencional de 3 dimensiones del tumor). Las mediciones se realizaron en el momento del diagnóstico y después de la quimioterapia de inducción antes de la resección del tumor primario. Ninguno de los métodos de medición de la respuesta del tumor primario al final de la quimioterapia de inducción predijo la supervivencia.[
Concentraciones de lactato–deshidrogenasa y ferritina
Las concentraciones séricas altas de lactato–deshidrogenasa (LDH) y ferritina produjeron tasas peores de SSC y de SG a 5 años en una cohorte internacional grande de pacientes con diagnóstico de neuroblastoma (n > 8575) de 1990 a 2016. Las concentraciones séricas más altas de LDH y ferritina también dieron como resultado tasas peores de SSC y de SG a 3 años en pacientes con neuroblastoma de riesgo alto después de 2009. En un análisis multivariante ajustado por edad en el momento del diagnóstico, estado de MYCN y enfermedad en estadio 4 según el INSS, las concentraciones de LDH y ferritina conservaron capacidad pronóstica independiente (P < 0,0001).[
Aunque las concentraciones séricas de ferritina y LDH no se evaluaron de forma crítica en el sistema de clasificación INRG original, en un análisis subsecuente del INRG Data Commons se ha demostrado con claridad la significancia estadística independiente de estas concentraciones séricas en el pronóstico de todos los pacientes y de los pacientes de riesgo alto, incluso en el período transcurrido entre 2010 y 2016. Por lo tanto, se indicó que estos valores, fácilmente obtenibles mediante pruebas de laboratorio, se incorporaran al sistema de clasificación pronóstica del INRG.[
Referencias:
- Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29.
Also available onlineAlso available online . Last accessed August 21, 2023. - National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available onlineAvailable online . Last accessed August 23, 2024. - Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28.
Also available onlineAlso available online . Last accessed August 21, 2023. - Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute.
Available onlineAvailable online . Last accessed September 5, 2024. - Gurney JG, Ross JA, Wall DA, et al.: Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992. J Pediatr Hematol Oncol 19 (5): 428-32, 1997 Sep-Oct.
- United States Census Bureau: Age and Sex Composition in the United States: 2018. U.S. Census Bureau, 2018.
Available onlineAvailable online . Last accessed August 21, 2023. - Mahapatra S, Challagundla KB: Neuroblastoma. Treasure Island, FL: StatPearls Publishing LLC, 2022.
Available onlineAvailable online . Last accessed August 21, 2023. - Campbell K, Siegel DA, Umaretiya PJ, et al.: A comprehensive analysis of neuroblastoma incidence, survival, and racial and ethnic disparities from 2001 to 2019. Pediatr Blood Cancer 71 (1): e30732, 2024.
- London WB, Castleberry RP, Matthay KK, et al.: Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children's Oncology Group. J Clin Oncol 23 (27): 6459-65, 2005.
- Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations). National Cancer Institute, 2012.
Also available onlineAlso available online . Last accessed May 22, 2024. - Takeuchi LA, Hachitanda Y, Woods WG, et al.: Screening for neuroblastoma in North America. Preliminary results of a pathology review from the Quebec Project. Cancer 76 (11): 2363-71, 1995.
- Woods WG, Gao RN, Shuster JJ, et al.: Screening of infants and mortality due to neuroblastoma. N Engl J Med 346 (14): 1041-6, 2002.
- Schilling FH, Spix C, Berthold F, et al.: Neuroblastoma screening at one year of age. N Engl J Med 346 (14): 1047-53, 2002.
- Heck JE, Ritz B, Hung RJ, et al.: The epidemiology of neuroblastoma: a review. Paediatr Perinat Epidemiol 23 (2): 125-43, 2009.
- Mossé YP, Laudenslager M, Longo L, et al.: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455 (7215): 930-5, 2008.
- Mosse YP, Laudenslager M, Khazi D, et al.: Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 75 (4): 727-30, 2004.
- Raabe EH, Laudenslager M, Winter C, et al.: Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene 27 (4): 469-76, 2008.
- van Limpt V, Schramm A, van Lakeman A, et al.: The Phox2B homeobox gene is mutated in sporadic neuroblastomas. Oncogene 23 (57): 9280-8, 2004.
- Serra A, Häberle B, König IR, et al.: Rare occurrence of PHOX2b mutations in sporadic neuroblastomas. J Pediatr Hematol Oncol 30 (10): 728-32, 2008.
- Satgé D, Moore SW, Stiller CA, et al.: Abnormal constitutional karyotypes in patients with neuroblastoma: a report of four new cases and review of 47 others in the literature. Cancer Genet Cytogenet 147 (2): 89-98, 2003.
- Mosse Y, Greshock J, King A, et al.: Identification and high-resolution mapping of a constitutional 11q deletion in an infant with multifocal neuroblastoma. Lancet Oncol 4 (12): 769-71, 2003.
- Moroni I, Bedeschi F, Luksch R, et al.: Costello syndrome: a cancer predisposing syndrome? Clin Dysmorphol 9 (4): 265-8, 2000.
- Cotton JL, Williams RG: Noonan syndrome and neuroblastoma. Arch Pediatr Adolesc Med 149 (11): 1280-1, 1995.
- Gutmann DH, Ferner RE, Listernick RH, et al.: Neurofibromatosis type 1. Nat Rev Dis Primers 3: 17004, 2017.
- Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.
- Bougnères P, Pantalone L, Linglart A, et al.: Endocrine manifestations of the rapid-onset obesity with hypoventilation, hypothalamic, autonomic dysregulation, and neural tumor syndrome in childhood. J Clin Endocrinol Metab 93 (10): 3971-80, 2008.
- Maas SM, Vansenne F, Kadouch DJ, et al.: Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 170 (9): 2248-60, 2016.
- Witkowski L, Nichols KE, Jongmans M, et al.: Germline pathogenic SMARCA4 variants in neuroblastoma. J Med Genet 60 (10): 987-992, 2023.
- Kim J, Vaksman Z, Egolf LE, et al.: Germline pathogenic variants in neuroblastoma patients are enriched in BARD1 and predict worse survival. J Natl Cancer Inst 116 (1): 149-159, 2024.
- Tolbert VP, Coggins GE, Maris JM: Genetic susceptibility to neuroblastoma. Curr Opin Genet Dev 42: 81-90, 2017.
- Matser YAH, Verly IRN, van der Ham M, et al.: Optimising urinary catecholamine metabolite diagnostics for neuroblastoma. Pediatr Blood Cancer 70 (6): e30289, 2023.
- Kratz CP, Rapisuwon S, Reed H, et al.: Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet 157 (2): 83-9, 2011.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Citak C, Karadeniz C, Dalgic B, et al.: Intestinal lymphangiectasia as a first manifestation of neuroblastoma. Pediatr Blood Cancer 46 (1): 105-7, 2006.
- Bourdeaut F, de Carli E, Timsit S, et al.: VIP hypersecretion as primary or secondary syndrome in neuroblastoma: A retrospective study by the Société Française des Cancers de l'Enfant (SFCE). Pediatr Blood Cancer 52 (5): 585-90, 2009.
- Mahoney NR, Liu GT, Menacker SJ, et al.: Pediatric horner syndrome: etiologies and roles of imaging and urine studies to detect neuroblastoma and other responsible mass lesions. Am J Ophthalmol 142 (4): 651-9, 2006.
- Conte M, Parodi S, De Bernardi B, et al.: Neuroblastoma in adolescents: the Italian experience. Cancer 106 (6): 1409-17, 2006.
- Matthay KK, Blaes F, Hero B, et al.: Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer Lett 228 (1-2): 275-82, 2005.
- Rudnick E, Khakoo Y, Antunes NL, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: clinical outcome and antineuronal antibodies-a report from the Children's Cancer Group Study. Med Pediatr Oncol 36 (6): 612-22, 2001.
- Antunes NL, Khakoo Y, Matthay KK, et al.: Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol 22 (4): 315-20, 2000 Jul-Aug.
- Pang KK, de Sousa C, Lang B, et al.: A prospective study of the presentation and management of dancing eye syndrome/opsoclonus-myoclonus syndrome in the United Kingdom. Eur J Paediatr Neurol 14 (2): 156-61, 2010.
- Pranzatelli MR: The neurobiology of the opsoclonus-myoclonus syndrome. Clin Neuropharmacol 15 (3): 186-228, 1992.
- Mitchell WG, Davalos-Gonzalez Y, Brumm VL, et al.: Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and neurologic sequelae. Pediatrics 109 (1): 86-98, 2002.
- Cooper R, Khakoo Y, Matthay KK, et al.: Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: histopathologic features-a report from the Children's Cancer Group. Med Pediatr Oncol 36 (6): 623-9, 2001.
- Pranzatelli MR, Tate ED, McGee NR: Demographic, Clinical, and Immunologic Features of 389 Children with Opsoclonus-Myoclonus Syndrome: A Cross-sectional Study. Front Neurol 8: 468, 2017.
- Hero B, Clement N, Øra I, et al.: Genomic Profiles of Neuroblastoma Associated With Opsoclonus Myoclonus Syndrome. J Pediatr Hematol Oncol 40 (2): 93-98, 2018.
- Catsman-Berrevoets CE, Aarsen FK, van Hemsbergen ML, et al.: Improvement of neurological status and quality of life in children with opsoclonus myoclonus syndrome at long-term follow-up. Pediatr Blood Cancer 53 (6): 1048-53, 2009.
- Connolly AM, Pestronk A, Mehta S, et al.: Serum autoantibodies in childhood opsoclonus-myoclonus syndrome: an analysis of antigenic targets in neural tissues. J Pediatr 130 (6): 878-84, 1997.
- Bell J, Moran C, Blatt J: Response to rituximab in a child with neuroblastoma and opsoclonus-myoclonus. Pediatr Blood Cancer 50 (2): 370-1, 2008.
- Corapcioglu F, Mutlu H, Kara B, et al.: Response to rituximab and prednisolone for opsoclonus-myoclonus-ataxia syndrome in a child with ganglioneuroblastoma. Pediatr Hematol Oncol 25 (8): 756-61, 2008.
- Pranzatelli MR, Tate ED, Travelstead AL, et al.: Rituximab (anti-CD20) adjunctive therapy for opsoclonus-myoclonus syndrome. J Pediatr Hematol Oncol 28 (9): 585-93, 2006.
- Ertle F, Behnisch W, Al Mulla NA, et al.: Treatment of neuroblastoma-related opsoclonus-myoclonus-ataxia syndrome with high-dose dexamethasone pulses. Pediatr Blood Cancer 50 (3): 683-7, 2008.
- Pranzatelli MR, Tate ED: Dexamethasone, Intravenous Immunoglobulin, and Rituximab Combination Immunotherapy for Pediatric Opsoclonus-Myoclonus Syndrome. Pediatr Neurol 73: 48-56, 2017.
- de Alarcon PA, Matthay KK, London WB, et al.: Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Health 2 (1): 25-34, 2018.
- Vik TA, Pfluger T, Kadota R, et al.: (123)I-mIBG scintigraphy in patients with known or suspected neuroblastoma: Results from a prospective multicenter trial. Pediatr Blood Cancer 52 (7): 784-90, 2009.
- Yang J, Codreanu I, Servaes S, et al.: I-131 MIBG post-therapy scan is more sensitive than I-123 MIBG pretherapy scan in the evaluation of metastatic neuroblastoma. Nucl Med Commun 33 (11): 1134-7, 2012.
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.
- Jennings RW, LaQuaglia MP, Leong K, et al.: Fetal neuroblastoma: prenatal diagnosis and natural history. J Pediatr Surg 28 (9): 1168-74, 1993.
- Brodeur GM: Spontaneous regression of neuroblastoma. Cell Tissue Res 372 (2): 277-286, 2018.
- Guan J, Hallberg B, Palmer RH: Chromosome Imbalances in Neuroblastoma-Recent Molecular Insight into Chromosome 1p-deletion, 2p-gain, and 11q-deletion Identifies New Friends and Foes for the Future. Cancers (Basel) 13 (23): , 2021.
- Schneiderman J, London WB, Brodeur GM, et al.: Clinical significance of MYCN amplification and ploidy in favorable-stage neuroblastoma: a report from the Children's Oncology Group. J Clin Oncol 26 (6): 913-8, 2008.
- Hiyama E, Hiyama K, Yokoyama T, et al.: Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1 (3): 249-55, 1995.
- Kitanaka C, Kato K, Ijiri R, et al.: Increased Ras expression and caspase-independent neuroblastoma cell death: possible mechanism of spontaneous neuroblastoma regression. J Natl Cancer Inst 94 (5): 358-68, 2002.
- Brodeur GM, Minturn JE, Ho R, et al.: Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res 15 (10): 3244-50, 2009.
- Yamamoto K, Ohta S, Ito E, et al.: Marginal decrease in mortality and marked increase in incidence as a result of neuroblastoma screening at 6 months of age: cohort study in seven prefectures in Japan. J Clin Oncol 20 (5): 1209-14, 2002.
- Okazaki T, Kohno S, Mimaya J, et al.: Neuroblastoma detected by mass screening: the Tumor Board's role in its treatment. Pediatr Surg Int 20 (1): 27-32, 2004.
- Fritsch P, Kerbl R, Lackner H, et al.: "Wait and see" strategy in localized neuroblastoma in infants: an option not only for cases detected by mass screening. Pediatr Blood Cancer 43 (6): 679-82, 2004.
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009.
Also available onlineAlso available online . Last accessed August 21, 2023. - Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.
- McCarthy LC, Chastain K, Flatt TG, et al.: Neuroblastoma in Adolescents and Children Older than 10 Years: Unusual Clinicopathologic and Biologic Features. J Pediatr Hematol Oncol 41 (8): 586-595, 2019.
- Mazzocco K, Defferrari R, Sementa AR, et al.: Genetic abnormalities in adolescents and young adults with neuroblastoma: A report from the Italian Neuroblastoma group. Pediatr Blood Cancer 62 (10): 1725-32, 2015.
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.
- Mossé YP, Deyell RJ, Berthold F, et al.: Neuroblastoma in older children, adolescents and young adults: a report from the International Neuroblastoma Risk Group project. Pediatr Blood Cancer 61 (4): 627-35, 2014.
- Kushner BH, Kramer K, LaQuaglia MP, et al.: Neuroblastoma in adolescents and adults: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 41 (6): 508-15, 2003.
- Suzuki M, Kushner BH, Kramer K, et al.: Treatment and outcome of adult-onset neuroblastoma. Int J Cancer 143 (5): 1249-1258, 2018.
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.
- Kubota M, Suita S, Tajiri T, et al.: Analysis of the prognostic factors relating to better clinical outcome in ganglioneuroblastoma. J Pediatr Surg 35 (1): 92-5, 2000.
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.
- Isaacs H: Fetal and neonatal neuroblastoma: retrospective review of 271 cases. Fetal Pediatr Pathol 26 (4): 177-84, 2007 Jul-Aug.
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.
- Vo KT, Matthay KK, Neuhaus J, et al.: Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol 32 (28): 3169-76, 2014.
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.
- Hiyama E, Yokoyama T, Hiyama K, et al.: Multifocal neuroblastoma: biologic behavior and surgical aspects. Cancer 88 (8): 1955-63, 2000.
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.
- Campbell K, Gastier-Foster JM, Mann M, et al.: Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children's Oncology Group. Cancer 123 (21): 4224-4235, 2017.
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.
- Burchill SA, Lewis IJ, Abrams KR, et al.: Circulating neuroblastoma cells detected by reverse transcriptase polymerase chain reaction for tyrosine hydroxylase mRNA are an independent poor prognostic indicator in stage 4 neuroblastoma in children over 1 year. J Clin Oncol 19 (6): 1795-801, 2001.
- Seeger RC, Reynolds CP, Gallego R, et al.: Quantitative tumor cell content of bone marrow and blood as a predictor of outcome in stage IV neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 18 (24): 4067-76, 2000.
- Bochennek K, Esser R, Lehrnbecher T, et al.: Impact of minimal residual disease detection prior to autologous stem cell transplantation for post-transplant outcome in high risk neuroblastoma. Klin Padiatr 224 (3): 139-42, 2012.
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.
- Streby KA, Parisi MT, Shulkin BL, et al.: Impact of diagnostic and end-of-induction Curie scores with tandem high-dose chemotherapy and autologous transplants for metastatic high-risk neuroblastoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 70 (8): e30418, 2023.
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.
- George RE, Perez-Atayde AR, Yao X, et al.: Tumor histology during induction therapy in patients with high-risk neuroblastoma. Pediatr Blood Cancer 59 (3): 506-10, 2012.
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.
- Moroz V, Machin D, Hero B, et al.: The prognostic strength of serum LDH and serum ferritin in children with neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 67 (8): e28359, 2020.
Características genómicas y biológicas del neuroblastoma
Características moleculares del neuroblastoma
Los niños con neuroblastoma se agrupan en subconjuntos con diferentes riesgos previstos de recaída de acuerdo con factores clínicos y marcadores biológicos presentes en el momento del diagnóstico.
- Pacientes con neuroblastoma de riesgo bajo o intermedio. Los pacientes clasificados con riesgo bajo o riesgo intermedio tienen un pronóstico favorable con tasas de supervivencia superiores al 95 %. El neuroblastoma de riesgo bajo o intermedio por lo general se presenta en niños menores de 18 meses. A menudo, estos tumores tienen ganancia de cromosomas enteros y son hiperdiploides cuando se los examina con citometría de flujo.[
11 ,22 ] - Pacientes con neuroblastoma de riesgo alto. El pronóstico de los pacientes con neuroblastoma de riesgo alto es más reservado con una tasa de supervivencia a largo plazo menor al 50 %. El neuroblastoma de riesgo alto suele presentarse en niños mayores de 18 meses, a menudo, con metástasis en hueso y médula ósea. En estos tumores, es habitual que se detecten anormalidades cromosómicas segmentarias (ganancias o pérdidas) o amplificación del gen MYCN. Según se observa en la medición con citometría de flujo, son casi diploides o casi tetraploides.[
11 ,22 ,33 ,44 ,55 ,66 ,77 ] Los tumores de riesgo alto pocas veces exhiben variantes exónicas, pero la mayoría de los tumores de riesgo alto carecen de dichas variantes génicas. Para obtener más información, consultar la sección Variantes exónicas en el neuroblastoma.
Las características genómicas clave del neuroblastoma de riesgo alto se describen a continuación:
- Anomalías cromosómicas segmentarias.
- Amplificaciones del gen MYCN.
- Activación de FOXR2.
- Tasas bajas de variantes exónicas; la alteración recurrente más común son las variantes activadoras de ALK.
- Alteraciones genómicas que promueven el mantenimiento de los telómeros.
Anomalías cromosómicas segmentarias
Las anomalías cromosómicas segmentarias, que a menudo se encuentran en 1p, 2p, 1q, 3p, 11q, 14q y 17p, se detectan mejor mediante hibridación genómica comparativa. Estas anomalías se encuentran en la mayoría de los tumores de neuroblastoma en estadio 4 o de riesgo alto.[
- Edad avanzada en el momento del diagnóstico.
- Estadio avanzado de la enfermedad.
- Riesgo más alto de recaída.
- Desenlace más precario.
En un análisis del neuroblastoma localizado, resecable y sin amplificación de MYCN, se evaluaron casos de dos estudios europeos consecutivos y una cohorte de Norte América (que incluyó casos en estadios 1, 2A y 2B de acuerdo al INSS) para detectar alteraciones cromosómicas segmentarias (a saber, ganancia de 1q, 2p y 17q, además de pérdida de 1p, 3p, 4p y 11q). En el estudio se descubrió que las características genómicas del tumor tenían una repercusión pronóstica diferente según la edad del paciente (<18 meses o >18 meses). Los pacientes se trataron solo con intervención quirúrgica, con independencia de la presencia de residuo tumoral.[
- La presencia de anomalías cromosómicas segmentarias, en particular la pérdida de 11q, redujo significativamente la supervivencia en los pacientes mayores de 18 meses con neuroblastoma en estadio 2, pero no en la cohorte de pacientes menores de 18 meses.
- La pérdida del cromosoma 1p es un factor de riesgo para la recaída, pero no para la disminución de la supervivencia general (SG) en los pacientes menores de 18 meses. La tasa de supervivencia sin complicaciones (SSC) a 5 años fue del 62 % en los pacientes con pérdida de 1p y del 87 % en aquellos sin pérdida de 1p (P = 0,019). La tasa de SG a 5 años fue del 92 % en los pacientes con pérdida de 1p y del 97 % en los pacientes sin pérdida de 1p.
- Las anomalías cromosómicas segmentarias (en particular, la pérdida de 11q) representan factores de riesgo para la reducción de la SSC y la SG en pacientes mayores de 18 meses. En los pacientes menores de 18 meses, solo las anomalías cromosómicas segmentarias condujeron a recaída y muerte; la pérdida de 11q fue el marcador más fuerte (pérdida de 11q: tasa de SSC a 5 años, 48 %; sin pérdida de 11q: tasa de SSC a 5 años, 85 %; P = 0,033; pérdida de 11q: tasa de SG a 5 años, 46 %; sin pérdida de 11q: tasa de SG a 5 años, 92 %; P = 0,038).
En un estudio de niños mayores de 12 meses con neuroblastomas primarios irresecables sin metástasis, se encontraron anomalías cromosómicas segmentarias en la mayoría de los pacientes. Los niños mayores fueron más propensos a presentarlas y tener más de estas anomalías por célula tumoral. En los niños de 12 a 18 meses, la presencia de anomalías cromosómicas segmentarias tuvo un efecto significativo en la SSC, pero no en la SG. Sin embargo, en los niños mayores de 18 meses, hubo una diferencia significativa en la SG entre aquellos con anomalías cromosómicas segmentarias (67 %) y aquellos sin anomalías cromosómicas segmentarias (100 %), con independencia de las características histológicas del tumor.[
Las anomalías cromosómicas segmentarias también permiten pronosticar la recidiva en lactantes con neuroblastoma metastásico o neuroblastoma localizado irresecable sin amplificación del gen MYCN.[
En un análisis de pacientes de riesgo intermedio en un estudio del Children's Oncology Group (COG), la pérdida de 11q, pero sin pérdida de 1p, se asoció a una SSC inferior; sin embargo, no afectó la SG (con pérdida de 11q y sin pérdida de 11q: tasas de SSC a 3 años, 68 % y 85 %, respectivamente P = 0,022; tasas de SG a 3 años, 88 % y 94 %, respectivamente; P = 0,09).[
En un análisis multivariante de 407 pacientes de 4 ensayos consecutivos del COG sobre alto riesgo, se observó que la pérdida de heterocigosidad de 11q es un factor de predicción significativo para la enfermedad progresiva, y que la ausencia de heterocigosidad de 11q se asociaba con tasas más altas de respuesta completa al final de la inducción y respuesta parcial al final de la inducción.[
En un estudio de colaboración internacional de 556 pacientes con neuroblastoma de riesgo alto se identificaron dos tipos de anomalías segmentarias en el número de copias que se relacionaron con desenlaces muy desfavorables. Se encontraron pérdidas distales de 6q en el 6 % de los pacientes y se relacionaron con una tasa de supervivencia a 10 años de solo el 3,4 %. Además de la amplificación de MYCN, se detectaron amplificaciones de regiones fuera del locus de MYCN en el 18 % de los pacientes y se relacionaron con una tasa de supervivencia a 10 años del 5,8 %.[
Amplificación del genMYCN
La amplificación de MYCN se detecta en el 16 % al 25 % de los tumores de neuroblastoma.[
De acuerdo a la mayoría de los análisis multivariantes de regresión de factores pronósticos, en todos los estadios de la enfermedad, la amplificación del gen MYCN permite predecir claramente un pronóstico más precario, tanto para el tiempo hasta la progresión del tumor como para la SG.[
En la cohorte de tumores localizados con amplificación de MYCN, los pacientes con tumores hiperdiploides tienen mejores desenlaces que aquellos con tumores diploides.[
Las características clínicas y biopatológicas más desfavorables se relacionan, en cierta medida, con la amplificación de MYCN. En un análisis multivariante de regresión logística en 7102 pacientes del estudio del Internacional Neuroblastoma Risk Group (INRG), las anomalías cromosómicas segmentarias agrupadas y las ganancias de 17q fueron las únicas características de pronóstico precario, aún cuando no estaban asociadas a la amplificación de MYCN. No obstante, las anomalías cromosómicas segmentarias en 11q, otra característica de pronóstico precario, son casi mutuamente excluyentes con la amplificación de MYCN.[
En una cohorte de 6223 pacientes de la base de datos del INRG con estado de MYCN conocido, el cociente de riesgos instantáneos (CRI) para la SG relacionada con la amplificación de MYCN fue de 6,3 (intervalo de confianza [IC] 95 %, 5,7–7,0; P < 0,001). El mayor efecto pronóstico adverso de la amplificación de MYCN en la SG se observó en los pacientes más jóvenes (<18 meses: CRI, 19,6; ≥18 meses: CRI, 3,0). Los pacientes cuyo desenlace se vio más afectado por el estado de MYCN fueron los que presentaban características favorables, como una edad menor de 18 meses, índice de mitosis cariorrexis alto y ferritina baja.[
La amplificación intratumoral heterogénea de MYCN (hetMNA) se refiere a la coexistencia de células tumorales con amplificación de MYCN (agrupadas o dispersas) y células tumorales sin amplificación de MYCN. La HetMNA se ha notificado de manera infrecuente. Es posible que se presente dentro del tumor, así como entre el tumor y la metástasis; al mismo tiempo o de forma transitoria durante la evolución de la enfermedad. El grupo de biología de la International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) investigó la importancia pronóstica de este subtipo de neuroblastoma. Se analizó el tejido tumoral de 99 pacientes en quienes se identificó hetMNA y que recibieron el diagnóstico entre 1991 y 2015, para aclarar la importancia pronóstica de los clones con amplificación de MYCN en casos de neuroblastoma sin amplificación de MYCN. Los pacientes menores de 18 meses presentaron un desenlace más favorable en todos los estadios en comparación con los pacientes de más edad. Se estableció una correlación significativa de los antecedentes genómicos con la frecuencia de la recaída y la SG. No se presentaron recaídas en los casos que solo presentaban anomalías cromosómicas numéricas. Este estudio indica que los tumores con hetMNA se evalúan teniendo en cuenta las características genómicas del tumor y el cuadro clínico, incluso la edad del paciente y el estadio de la enfermedad. Se necesitan más estudios en pacientes menores de 18 meses que exhiban enfermedad localizada con hetMNA.[
Activación deFOXR2
La expresión del gen FOXR2 se observa en alrededor del 8 % de los casos de neuroblastoma. La expresión del gen FOXR2 por lo general es nula después del nacimiento, a excepción de los tejidos reproductivos en varones.[
El neuroblastoma con activación de FOXR2 se observa en tasas similares en los casos con riesgo alto y sin riesgo alto.[
Variantes exónicas en el neuroblastoma (incluso variantes y amplificaciones deALK)
En comparación con los cánceres en adultos, el neuroblastoma infantil exhibe un número bajo de variantes por genoma que afectan la secuencia de proteínas (10–20 por genoma).[
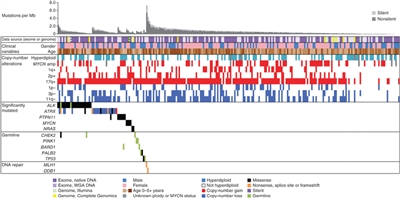
El gen ALK proporciona las instrucciones para la elaboración de un receptor tirosina–cinasa de superficie celular que se expresa en concentraciones importantes solo en los encéfalos embrionarios y neonatales en desarrollo. La variante exónica de ALK es la alteración que se encuentra con más frecuencia en el neuroblastoma. Las variantes germinales de ALK se identificaron como la causa principal del neuroblastoma hereditario. Se encontró que las variantes exónicas activadoras de ALK adquiridas de forma somática también son oncoiniciadoras del neuroblastoma.[
En 2 estudios de cohortes numerosas se examinaron los marcadores clínicos y la importancia pronóstica de las alteraciones en ALK. En un estudio del COG se analizó el estado de ALK en 1596 muestras de diagnóstico de neuroblastoma en todos los grupos de riesgo.[
- Las variantes de ALK que afectan el dominio de tirosina–cinasa se presentaron sobre todo en 3 puntos de gran actividad (posiciones F1174, R1275 y F1245); el 10 % al 15 % de las variantes ocurrieron en otras posiciones del dominio de cinasa.
- En la cohorte del COG la frecuencia de las variantes de ALK fue del 10 % en el grupo de neuroblastoma de riesgo alto, del 8 % en el grupo del neuroblastoma de riesgo intermedio y del 6 % en el grupo de neuroblastoma de riesgo bajo.
- En la población de riesgo alto de la SIOPEN, las variantes de ALK se dividieron en clonales (frecuencia de la variante del alelo [VAF] >20 %) y subclonales (VAF 0,1–20 %). Se observaron variantes clonales de ALK en un 10 % de los casos, y variantes subclonales en un 3,9 % de los pacientes. El 13,9 % de los casos presentaban variantes de ALK.
- Se encontraron tasas más altas de variantes de ALK en los pacientes con tumores con amplificación de MYCN en comparación con aquellos sin amplificación de MYCN: el 10,9 % versus el 7,2 %, respectivamente, en la cohorte del COG y el 14 % versus el 6,5 %, respectivamente, en la cohorte de la SIOPEN (para las variantes clonales de ALK).
- En los pacientes con neuroblastoma de riesgo alto, la amplificación de ALK se observó en cerca del 4 % de los casos en las cohortes del COG y de la SIOPEN. La amplificación de ALK se presentó casi de manera exclusiva en casos que también tenían amplificación de MYCN.
- Las alteraciones en ALK se relacionaron con un pronóstico inferior en los pacientes con neuroblastoma de riesgo alto en el estudio del COG y en el de la SIOPEN:
- En la cohorte de la SIOPEN, se observó una diferencia estadísticamente significativa en la SG entre los casos con amplificación de ALK (ALKa) o variante clonal de ALK (ALKm) versus ALKm subclonal o sin alteraciones en ALK (tasa de SG a 5 años: ALKa, 26 % [IC 95 %, 10–47 %]; ALKm clonal, 33 % [IC 95 %, 21–44 %]; ALKm subclonal, 48 % [IC 95 %, 26–67 %]; y sin alteración, 51 % [IC 95 %, 46–55 %], respectivamente P = 0,001). En un modelo multivariante, la amplificación en ALK (CRI, 2,38; P = 0,004) y la variante clonal de ALK (CRI, 1,77; P = 0,001) fueron factores independientes de predicción de desenlace precario.
- En la población de neuroblastoma de riesgo alto del COG se observaron pronósticos inferiores, similares a los observados en la cohorte de la SIOPEN, en los casos con variantes de ALK y amplificaciones en ALK.
En un estudio donde se compararon los datos genómicos de neuroblastomas de diagnóstico primario que se originaron en la glándula suprarrenal (n = 646) con los de neuroblastomas originados en los ganglios simpáticos torácicos (n = 118), el 16 % de los tumores torácicos albergaban variantes de ALK.[
Los inhibidores de molécula pequeña de la cinasa de ALK, como el loratinib (añadido a la terapia convencional), se están probando en pacientes de neuroblastoma con variante recurrente de ALK (NCT03107988) y en pacientes con neuroblastoma de riesgo alto recién diagnosticado con activación de ALK (COG ANBL1531).[
Evolución genómica de las variantes exónicas
Hay pocos datos sobre la evolución genómica de las variantes exónicas desde el diagnóstico hasta la recaída del neuroblastoma. Se aplicó la secuenciación del genoma completo a 23 muestras emparejadas de tumores de neuroblastoma obtenidas en el momento del diagnóstico y de la recaída con el fin de definir las alteraciones genéticas somáticas relacionadas con la recaída;[
- En el primer estudio, se encontró una mayor incidencia de variantes de los genes relacionados con la señalización de RAS-MAPK en tumores de recaída en comparación con los tumores del mismo paciente en el momento del diagnóstico: 15 de 23 muestras de recaída contenían variantes somáticas de los genes que participan en esta vía; además, cada variante fue compatible con la activación de la vía.[
3333 ]Asimismo, en 3 muestras de recaída se encontraron alteraciones estructurales que comprometían genes de la vía MAPK compatibles con activación de la vía, es decir que se detectaron anomalías en esta vía en 18 de 23 (78 %) muestras de recaída. Se encontraron anomalías en ALK (n = 10) y en NF1 (n = 2), además de una anomalía en cada uno de los siguientes genes: NRAS, KRAS, HRAS, BRAF, PTPN11 y FGFR1. Incluso con una secuenciación masiva, 7 de las 18 alteraciones no fueron detectables en el tumor primario, esto subraya la evolución de las variantes que presumiblemente conducen a la recaída y la importancia de las evaluaciones genómicas de los tejidos obtenidos en el momento de la recaída.
- En el segundo estudio, no se observaron variantes de ALK en el momento del diagnóstico ni en la recaída, pero se detectaron variantes de un solo nucleótido recurrentes específicas de la recaída en 11 genes, incluso el presunto gen supresor de tumores del neuroblastoma CHD5 localizado en el cromosoma 1p36.[
3434 ] - En un tercer estudio retrospectivo de secuenciación de variantes, se usaron datos de la Foundation Medicine para comparar las muestras de tumores de pacientes con neuroblastoma recién diagnosticado con las muestras de tumores de pacientes de neuroblastoma resistente al tratamiento y en recaída. En el estudio se encontró un porcentaje más alto de variantes susceptibles de tratamiento dirigido con fármacos actuales en el grupo de pacientes en recaída y resistente al tratamiento.[
3535 ] - En un cuarto estudio se evaluó la frecuencia de las alteraciones en ALK en el momento del diagnóstico y de la recaída. Se presentaron tasas significativamente más altas de variantes de ALK en la recaída que en el diagnóstico (17,7 % en la recaída vs. 10,5 % en el diagnóstico). La tasa de amplificaciones en ALK no fue diferente entre el momento del diagnóstico y de la recaída.[
3636 ]
Dada la naturaleza metastásica generalizada del neuroblastoma de riesgo alto y en recaída, el uso de métodos para medir el DNA tumoral circulante (ctDNA) quizás revele otras alteraciones genómicas que no se encuentran con las biopsias tumorales convencionales. Además, estos abordajes han demostrado la capacidad para detectar variantes de resistencia en pacientes con neuroblastoma tratados con inhibidores de ALK.[
En un estudio de secuenciación masiva, 276 muestras de neuroblastoma (en todos los estadios y de pacientes de todas las edades en el momento del diagnóstico) se evaluaron mediante secuenciación de solo 2 puntos de gran actividad de variantes de ALK, lo que reveló un 4,8 % de variantes clonales y otro 5 % de variantes subclonales. Este hallazgo indica que las variantes subclonales del gen ALK son comunes.[
Alteraciones genómicas que promueven el mantenimiento de los telómeros
El alargamiento de los telómeros, los extremos de los cromosomas, promueve la supervivencia celular. Por otra parte, los telómeros se acortan con cada replicación celular, lo que resulta finalmente en la incapacidad de la célula para replicarse. Los pacientes cuyos tumores carecen de mecanismos de mantenimiento de telómeros tienen un pronóstico excelente, mientras que los que albergan mecanismos de mantenimiento de telómeros tienen un pronóstico bastante más precario.[
- Los reordenamientos cromosómicos que comprometen una región cromosómica en 5p15.33 próxima al gen TERT, que codifica la unidad catalítica de la telomerasa, se presentan en el 20 % al 25 % de los casos de neuroblastoma de riesgo alto y son mutuamente excluyentes con las amplificaciones de MYCN y la activación del alargamiento alternativo de los telómeros.[
2424 ,2525 ,4141 ] Los reordenamientos inducen el aumento regulado de la transcripción de TERT al yuxtaponer la secuencia codificante de TERT con fuertes elementos potenciadores. Los niños cuyos tumores presentan reordenamientos en TERT tienen un pronóstico precario, comparable con el pronóstico de los niños con tumores con amplificación de MYCN.[4141 ] Es posible usar la secuenciación de última generación o la hibridación fluorescente in situ (FISH) para identificar estas alteraciones. En un estudio se identificaron reordenamientos en TERT mediante FISH en el 6 % de todos los pacientes con tumores neuroblásticos, con independencia del grupo de riesgo, y en el 12,4 % de los pacientes con tumores neuroblásticos de riesgo alto.[4242 ] - Otro mecanismo que promueve la sobreexpresión de TERT es la amplificación de MYCN,[
4343 ] que se relaciona con cerca del 40 % al 50 % de los casos de neuroblastoma de riesgo alto. - El alargamiento alternativo de los telómeros es un mecanismo adicional de mantenimiento de telómeros que utilizan los tumores de neuroblastoma. La activación del alargamiento alternativo de los telómeros se presenta en alrededor del 20 % al 25 % de los casos de riesgo alto recién diagnosticados, en comparación con cerca del 5 % al 12 % de los casos de riesgo bajo y riesgo intermedio.[
4141 ,4444 ,4545 ] En comparación con los casos recién diagnosticados, la proporción de casos de neuroblastoma con tumores positivos para el alargamiento alternativo de los telómeros fue mayor en una cohorte de pacientes en recaída (10 % vs. 48 %, respectivamente). Es posible que este hallazgo refleje una evolución relativamente indolente de los tumores con activación del alargamiento alternativo de los telómeros tras la recaída, en comparación con la evolución clínica de otros tumores después de la recaída. Con el tiempo, la proporción de pacientes con neuroblastomas para el alargamiento alternativo de los telómeros en recaída (entre los pacientes con neuroblastoma) parece mayor que la de pacientes con otro tipo de tumor que recayeron (entre los pacientes diagnosticados con ese tumor).[4444 ] Los casos de neuroblastoma con activación del alargamiento alternativo de los telómeros tienen expresión baja de TERT y se pueden identificar mediante prueba inmunohistoquímica por la presencia de cuerpos nucleares de leucemia promielocítica asociados a alargamiento alternativo de los telómeros. Estos casos también se identifican con una prueba de círculo C o con FISH al usar una sonda telomérica para observar los puntos brillantes de los telómeros.[4444 ,4545 ] Alrededor del 55 % al 60 % de los casos positivos para el alargamiento alternativo de los telómeros se caracterizan por presentar variantes deletéreas de ATRX.[2626 ,4444 ,4545 ] Los casos que no tienen variantes de ATRX a menudo presentan expresión baja de la proteína ATRX.[4444 ]Los tumores positivos para el alargamiento alternativo de los telómeros en las poblaciones infantiles casi nunca se presentan antes de los 18 meses de edad y surgen casi de forma exclusiva en niños más grandes (mediana de edad en el momento del diagnóstico, cerca de los 8 años).[
4141 ,4444 ] La proporción de casos de neuroblastoma con variantes de ATRX aumenta con la edad en las poblaciones de adolescentes y adultos jóvenes.[2626 ]El pronóstico para la SSC de los pacientes de riesgo alto con activación del alargamiento alternativo de los telómeros es tan precario como el de los pacientes con amplificación de MYCN,[
4141 ,4444 ] sin embargo, la SG es superior en los pacientes con activación del alargamiento alternativo de los telómeros. La SG más favorable es consecuencia de una evolución un poco más prolongada de la enfermedad después de la recaída, pero con una supervivencia a largo plazo (10 a 15 años) tan baja como la de otros pacientes con neuroblastoma de riesgo alto.[4141 ,4444 ] En un informe, la SSC y la SG en los pacientes de riesgo intermedio y riesgo bajo con activación del alargamiento alternativo de los telómeros fueron similares a las observadas en pacientes positivos para el alargamiento alternativo de los telómeros con enfermedad de riesgo alto.[4444 ]
Otros factores biológicos relacionados con el pronóstico
Expresión de MYC y MYCN
Con la inmunotinción de las proteínas MYC y MYCN en un subconjunto restringido de 357 tumores de neuroblastoma indiferenciado o poco diferenciado, se demostró que la expresión elevada de las proteínas MYC o MYCN es un factor pronóstico importante.[
- Los pacientes con tumores con características histológicas favorables sin expresión alta de MYCN o MYC tuvieron una supervivencia favorable (SSC a 3 años, 89,7 ± 5,5 %; SG a 3 años, 97 ± 3,2 %).
- Los pacientes con tumores con características histológicas indiferenciadas o poco diferenciadas sin expresión de MYCN o MYC tuvieron una tasa de SSC a 3 años del 63,1 % (± 13,6 %) y una tasa de SG a 3 años del 83,5 % (± 9,4 %).
- Las tasas de SSC a 3 años en pacientes con amplificación de MYCN, expresión alta de MYCN y expresión alta de MYC fueron del 48,1 % (± 11,5 %), del 46,2 % (± 12 %) y del 43,4 % (± 23,1 %), respectivamente. Las tasas de SG fueron del 65,8 % (± 11,1 %), del 63,2 % (± 12,1 %), y del 63,5 % (± 19,2 %), respectivamente.
- Además, cuando se hizo un análisis multivariante de la expresión alta de las proteínas MYC y MYCN y otros factores pronósticos, como la amplificación génica de MYC/MYCN, se concluyó que la expresión alta de las proteínas MYC y MYCN es independiente de otros marcadores pronósticos.
Cinasas receptoras de neurotrofina
La expresión de cinasas receptoras de neurotrofina y sus ligandos varía entre los tumores de riesgo alto y de riesgo bajo. El receptor TrkA se encuentra en tumores de riesgo bajo y se postula que la ausencia de su ligando NGF produce la remisión espontánea del tumor. En contraste, el receptor TrkB se encuentra en los tumores de riesgo alto que también expresan su ligando, BDNF, que promueve la proliferación y la supervivencia celular del neuroblastoma.[
Referencias:
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Schleiermacher G, Mosseri V, London WB, et al.: Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br J Cancer 107 (8): 1418-22, 2012.
- Janoueix-Lerosey I, Schleiermacher G, Michels E, et al.: Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol 27 (7): 1026-33, 2009.
- Schleiermacher G, Michon J, Ribeiro A, et al.: Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study). Br J Cancer 105 (12): 1940-8, 2011.
- Carén H, Kryh H, Nethander M, et al.: High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A 107 (9): 4323-8, 2010.
- Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al.: Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28 (19): 3122-30, 2010.
- Defferrari R, Mazzocco K, Ambros IM, et al.: Influence of segmental chromosome abnormalities on survival in children over the age of 12 months with unresectable localised peripheral neuroblastic tumours without MYCN amplification. Br J Cancer 112 (2): 290-5, 2015.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Ambros IM, Tonini GP, Pötschger U, et al.: Age Dependency of the Prognostic Impact of Tumor Genomics in Localized Resectable MYCN-Nonamplified Neuroblastomas. Report From the SIOPEN Biology Group on the LNESG Trials and a COG Validation Group. J Clin Oncol 38 (31): 3685-3697, 2020.
- Pinto N, Naranjo A, Ding X, et al.: Impact of Genomic and Clinical Factors on Outcome of Children ≥18 Months of Age with Stage 3 Neuroblastoma with Unfavorable Histology and without MYCN Amplification: A Children's Oncology Group (COG) Report. Clin Cancer Res 29 (8): 1546-1556, 2023.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.
- Depuydt P, Boeva V, Hocking TD, et al.: Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J Natl Cancer Inst 110 (10): 1084-1093, 2018.
- Ambros PF, Ambros IM, Brodeur GM, et al.: International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100 (9): 1471-82, 2009.
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.
- Plantaz D, Vandesompele J, Van Roy N, et al.: Comparative genomic hybridization (CGH) analysis of stage 4 neuroblastoma reveals high frequency of 11q deletion in tumors lacking MYCN amplification. Int J Cancer 91 (5): 680-6, 2001.
- Maris JM, Hogarty MD, Bagatell R, et al.: Neuroblastoma. Lancet 369 (9579): 2106-20, 2007.
- Campbell K, Shyr D, Bagatell R, et al.: Comprehensive evaluation of context dependence of the prognostic impact of MYCN amplification in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 66 (8): e27819, 2019.
- Berbegall AP, Bogen D, Pötschger U, et al.: Heterogeneous MYCN amplification in neuroblastoma: a SIOP Europe Neuroblastoma Study. Br J Cancer 118 (11): 1502-1512, 2018.
- Schmitt-Hoffner F, van Rijn S, Toprak UH, et al.: FOXR2 Stabilizes MYCN Protein and Identifies Non-MYCN-Amplified Neuroblastoma Patients With Unfavorable Outcome. J Clin Oncol 39 (29): 3217-3228, 2021.
- Sturm D, Orr BA, Toprak UH, et al.: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164 (5): 1060-72, 2016.
- Pugh TJ, Morozova O, Attiyeh EF, et al.: The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3): 279-84, 2013.
- Peifer M, Hertwig F, Roels F, et al.: Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526 (7575): 700-4, 2015.
- Valentijn LJ, Koster J, Zwijnenburg DA, et al.: TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47 (12): 1411-4, 2015.
- Cheung NK, Zhang J, Lu C, et al.: Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307 (10): 1062-71, 2012.
- Molenaar JJ, Koster J, Zwijnenburg DA, et al.: Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483 (7391): 589-93, 2012.
- Sausen M, Leary RJ, Jones S, et al.: Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45 (1): 12-7, 2013.
- Bresler SC, Weiser DA, Huwe PJ, et al.: ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 26 (5): 682-94, 2014.
- Janoueix-Lerosey I, Lequin D, Brugières L, et al.: Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455 (7215): 967-70, 2008.
- Bellini A, Pötschger U, Bernard V, et al.: Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol 39 (30): 3377-3390, 2021.
- Oldridge DA, Truong B, Russ D, et al.: Differences in Genomic Profiles and Outcomes Between Thoracic and Adrenal Neuroblastoma. J Natl Cancer Inst 111 (11): 1192-1201, 2019.
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.
- Rosswog C, Fassunke J, Ernst A, et al.: Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer 128 (8): 1559-1571, 2023.
- Bosse KR, Giudice AM, Lane MV, et al.: Serial Profiling of Circulating Tumor DNA Identifies Dynamic Evolution of Clinically Actionable Genomic Alterations in High-Risk Neuroblastoma. Cancer Discov 12 (12): 2800-2819, 2022.
- Berko ER, Witek GM, Matkar S, et al.: Circulating tumor DNA reveals mechanisms of lorlatinib resistance in patients with relapsed/refractory ALK-driven neuroblastoma. Nat Commun 14 (1): 2601, 2023.
- Bellini A, Bernard V, Leroy Q, et al.: Deep Sequencing Reveals Occurrence of Subclonal ALK Mutations in Neuroblastoma at Diagnosis. Clin Cancer Res 21 (21): 4913-21, 2015.
- Ackermann S, Cartolano M, Hero B, et al.: A mechanistic classification of clinical phenotypes in neuroblastoma. Science 362 (6419): 1165-1170, 2018.
- Roderwieser A, Sand F, Walter E, et al.: Telomerase is a prognostic marker of poor outcome and a therapeutic target in neuroblastoma. JCO Precis Oncol 3: 1-20, 2019.
- Yu Y, Zhang M, Yao X, et al.: Translational practice of fluorescence in situ hybridisation to identify neuroblastic tumours with TERT rearrangements. J Pathol Clin Res 9 (6): 475-487, 2023.
- Mac SM, D'Cunha CA, Farnham PJ: Direct recruitment of N-myc to target gene promoters. Mol Carcinog 29 (2): 76-86, 2000.
- Hartlieb SA, Sieverling L, Nadler-Holly M, et al.: Alternative lengthening of telomeres in childhood neuroblastoma from genome to proteome. Nat Commun 12 (1): 1269, 2021.
- Koneru B, Lopez G, Farooqi A, et al.: Telomere Maintenance Mechanisms Define Clinical Outcome in High-Risk Neuroblastoma. Cancer Res 80 (12): 2663-2675, 2020.
- Wang LL, Teshiba R, Ikegaki N, et al.: Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br J Cancer 113 (1): 57-63, 2015.
- Maris JM, Matthay KK: Molecular biology of neuroblastoma. J Clin Oncol 17 (7): 2264-79, 1999.
Clasificación de los tumores neuroblásticos
Los neuroblastomas se clasifican como uno de los tumores de células pequeñas, redondas y azules de la niñez. Conforman un grupo heterogéneo de tumores compuestos de agregados celulares con diferentes grados de diferenciación: ganglioneuromas maduros, ganglioneuroblastomas menos maduros y neuroblastomas inmaduros; ello refleja el potencial maligno variable de estos tumores.[
Los siguientes son los dos sistemas de clasificación del neuroblastoma:
-
Sistema de la International Neuroblastoma Pathology Classification (INPC)Sistema de la International Neuroblastoma Pathology Classification (INPC) . -
Sistema de la International Neuroblastoma Risk Group (INRG) ClassificationSistema de la International Neuroblastoma Risk Group (INRG) Classification .
Sistema de la Internacional Neuroblastoma Pathology Classification
El sistema de la Internacional Neuroblastoma Pathology Classification (INPC) derivó de la experiencia con la clasificación de Shimada original. En el
- Volumen de estroma schwanniano.
- Grado de maduración neuroblástica.
- Índice de mitosis-cariorrexis (MKI) de las células neuroblásticas.
Los pronósticos favorables y desfavorables se definen según estos parámetros histológicos y la edad del paciente. En varios estudios, se confirmó la importancia pronóstica de este sistema de clasificación y de los sistemas relacionados en los que se usan criterios similares (consultar el
| International Neuroblastoma Pathology Classification | Clasificación de Shimada original | Grupo pronóstico | |||
|---|---|---|---|---|---|
| MKI = índice de mitosis-cariorrexis. | |||||
| a Reproducción autorizada. Copyright © 1999 American Cancer Society. Todos los derechos reservados.[ |
|||||
| b Los subtipos de neuroblastoma se describen en detalle en otra parte.[ |
|||||
| c Subtipo poco frecuente, sobre todo cuando se diagnostica en este grupo de edad. Se necesita más investigación y análisis. | |||||
| d Los grupos pronósticos de estas categorías de tumores no se relacionan con la edad del paciente. | |||||
| Neuroblastoma: | (Estroma schwanniano escaso)b | Estroma escaso | |||
| Favorable: | Favorable | Favorable | |||
| <1,5 años | Tumor poco diferenciado o en diferenciación y tumor con MKI bajo o intermedio | ||||
| 1,5–5 años | Tumor en diferenciación y con MKI bajo | ||||
| Desfavorable: | Desfavorable | Desfavorable | |||
| <1,5 años | a) tumor indiferenciadoc | ||||
| b) tumor con MKI alto | |||||
| 1,5–5 años | a) tumor indiferenciado o poco diferenciado | ||||
| b) tumor con MKI intermedio o alto | |||||
| ≥5 años | Todos los tumores | ||||
| Ganglioneuroblastoma, entremezclado | (estroma schwanniano abundante) | Estroma abundante entremezclado (favorable) | Favorabled | ||
| Ganglioneuroma: | (estroma schwanniano dominante) | ||||
| En maduración | Bien diferenciado (favorable) | Favorabled | |||
| Maduro | Ganglioneuroma | ||||
| Ganglioneuroblastoma, nodular | (compuesto por mezcla de estroma schwanniano abundante, estroma dominante y estroma escaso) | Nodular con estroma abundante (desfavorable) | Desfavorabled | ||
La mayoría de los neuroblastomas con amplificación de MYCN tienen características histológicas de la INPC desfavorables, pero cerca del 7 % tienen características histológicas favorables. Por lo general, los tumores no expresan MYCN, a pesar de la amplificación de este gen; estos pacientes tienen un pronóstico más favorable que los pacientes cuyos tumores presentan amplificación de MYCN y sobreexpresión de MYCN.[
Se analizaron los componentes individuales de los datos de la INPC procedentes del INRG Data Commons (18 865 pacientes), y el análisis validó la capacidad pronóstica independiente de la edad en el momento del diagnóstico, la categoría histológica, el MKI y el grado de diferenciación. Se identificaron 4 grupos de pronóstico histológico relacionados con la edad (edad <18 meses con MKI bajo vs. MKI alto; edad >18 meses con tumores diferenciados vs. indiferenciados o poco diferenciados). Del mismo modo, mediante el uso de un esquema de riesgo carente de los factores de confusión de edad y de la INPC, en este análisis se identificó un nuevo y desfavorable subgrupo de pacientes mayores de 547 días de edad con tumores diploides en estadio 1 o 2, sin amplificación de MYCN o con MKI intermedio o alto, quienes tuvieron una tasa de supervivencia sin complicaciones (SSC) muy precaria (tasa de SSC, 46 %).[
Sistema de la International Neuroblastoma Risk Group Classification
En el sistema de la International Neuroblastoma Risk Group (INRG) Classification se utilizó un análisis del árbol de supervivencia para comparar 35 factores pronósticos en más de 8800 pacientes con neuroblastoma de varios de ensayos clínicos. En el análisis se incluyeron las siguientes características histológicas subyacentes de la INCP (sistema de Shimada):[
- Categoría diagnóstica.
- Grado de diferenciación.
- Índice de mitosis-cariorrexis (MKI).
Debido a que la edad del paciente se usa en todos los sistemas de estratificación del riesgo, era deseable un sistema de clasificación celular que no empleara la edad del paciente, por lo que se usaron los criterios histológicos subyacentes en el árbol de decisión final, en lugar de la INPC o la clasificación de Shimada. Los hallazgos histológicos permitieron diferenciar de forma más clara 2 subconjuntos de pacientes según los grupos pronósticos, conforme se muestra en el
| Estadio del INSS o subtipo histológico | Número de casos | SSC (%) | SG (%) | |
|---|---|---|---|---|
| SSC = supervivencia sin complicaciones; GN = ganglioneuroma; GNB = ganglioneuroblastoma; INSS = International Neuroblastoma Staging System; NB = neuroblastoma; SG = supervivencia general. | ||||
| a Adaptado de Cohn et al.[ |
||||
| Estadio 1, 2, 3, 4S del INSS | 5131 | 83 ± 1 | 91 ± 1 | |
| GN, en maduración | 162 | 97 ± 2 | 98 ± 2 | |
| GNB, entremezclado | ||||
| NB | 4970 | 83 ± 1 | 90 ± 1 | |
| GNB, nodular | ||||
| Estadio 2, 3 del INSS; edad >547 días | 260 | 69 ± 3 | 81 ± 2 | |
| 11q normal y en diferenciación | 16 | 80 ± 16 | 100 | |
| Anomalía de 11q o indiferenciado | 49 | 61 ± 11 | 73 ± 11 | |
Los subconjuntos histológicos del INRG se incorporan en el esquema de clasificación de riesgo del INRG. Para obtener más información, consultar el
Referencias:
- Joshi VV, Silverman JF: Pathology of neuroblastic tumors. Semin Diagn Pathol 11 (2): 107-17, 1994.
- Shimada H, Ambros IM, Dehner LP, et al.: The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86 (2): 364-72, 1999.
- Shimada H, Umehara S, Monobe Y, et al.: International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (9): 2451-61, 2001.
- Goto S, Umehara S, Gerbing RB, et al.: Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (10): 2699-708, 2001.
- Peuchmaur M, d'Amore ES, Joshi VV, et al.: Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 98 (10): 2274-81, 2003.
- Teshiba R, Kawano S, Wang LL, et al.: Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: a report from the Children's Oncology Group. Pediatr Dev Pathol 17 (6): 441-9, 2014 Nov-Dec.
- Shimada H, Ambros IM, Dehner LP, et al.: Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer 86 (2): 349-63, 1999.
- Suganuma R, Wang LL, Sano H, et al.: Peripheral neuroblastic tumors with genotype-phenotype discordance: a report from the Children's Oncology Group and the International Neuroblastoma Pathology Committee. Pediatr Blood Cancer 60 (3): 363-70, 2013.
- Sokol E, Desai AV, Applebaum MA, et al.: Age, Diagnostic Category, Tumor Grade, and Mitosis-Karyorrhexis Index Are Independently Prognostic in Neuroblastoma: An INRG Project. J Clin Oncol 38 (17): 1906-1918, 2020.
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Okamatsu C, London WB, Naranjo A, et al.: Clinicopathological characteristics of ganglioneuroma and ganglioneuroblastoma: a report from the CCG and COG. Pediatr Blood Cancer 53 (4): 563-9, 2009.
Información sobre los estadios del neuroblastoma
Evaluación para la estadificación
Alrededor del 70 % de los pacientes con neuroblastoma tienen enfermedad metastásica en el momento del diagnóstico. Se realiza una evaluación completa de la enfermedad metastásica antes de iniciar el tratamiento. Por lo general, se hacen los estudios descritos a continuación.[
Gammagrafía con metayodobencilguanidina
La extensión de la enfermedad metastásica se evalúa mediante una gammagrafía con metayodobencilguanidina (MlBG), que sirve para todos los sitios de enfermedad, incluso el tejido blando, la médula ósea y el hueso cortical. Alrededor del 90 % de los neuroblastomas estarán ávidos de MIBG. La gammagrafía con MIBG tiene una sensibilidad y especificidad del 90 % al 99 %, y la avidez de MIBG se distribuye por igual entre los sitios primarios y metastásicos.[
Las imágenes con 123I-MIBG son idóneas para identificar metástasis en el tejido blando y los huesos; en una comparación prospectiva, demostraron ser superiores al uso de la tomografía por emisión de positrones con tomografía computarizada (TEP-TC).[
Las gammagrafías iniciales con MIBG realizadas en el momento del diagnóstico son un método excelente para verificar la respuesta de la enfermedad y realizar la vigilancia posterior al tratamiento.[
Métodos de puntaje Curie y SIOPEN
Varios grupos han investigado un método semicuantitativo de puntaje para evaluar el alcance de la enfermedad y el valor pronóstico. Los métodos más comunes de puntaje que se utilizan para evaluar el alcance de la enfermedad y la respuesta son los métodos de Curie y de la Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN).
- Método de puntaje Curie: el puntaje Curie es un sistema de puntaje semicuantitativo formulado para predecir el alcance y la gravedad de la enfermedad ávida de MIBG. El uso del sistema de puntaje Curie fue evaluado como un marcador pronóstico de la respuesta y la supervivencia de neuroblastomas de riesgo alto recién diagnosticados, ávidos de MIBG en estadio 4 (N = 280), tratados con el protocolo del Children's Oncology Group (COG) COG-A3973 (NCT00004188). Para pacientes de neuroblastoma sin amplificación de MYCN, un puntaje Curie superior a 2 después de la quimioterapia de inducción identificó un riesgo más alto de un episodio, independiente de otros factores clínicos y biológicos conocidos del neuroblastoma, como edad, estado de MYCN, ploidía, índice de mitosis-cariorrexis y grado histológico.[
77 ] Para pacientes de tumores con amplificación de MYCN, un puntaje Curie superior a 0 después de la quimioterapia de inducción se relacionó con desenlaces más precarios.La importancia pronóstica de los puntajes Curie después de la inducción se validó en una cohorte independiente de pacientes.[
88 ] Se hizo un estudio retrospectivo de los puntajes Curie de gammagrafías con 123I-MIBG obtenidas en pacientes de riesgo alto inscritos de manera prospectiva en el ensayo SIOPEN/HR-NBL1 (NCT00030719). Se evaluaron gammagrafías de 10 regiones anatómicas; cada región fue calificada entre 0 y 3 según el alcance de la enfermedad y se generó un puntaje Curie acumulado. En el ensayo SIOPEN/HR-NBL1 se usó un puntaje Curie de 12 como valor límite con utilidad pronóstica óptima en el momento del diagnóstico, y se observó una diferencia significativa en el desenlace según el puntaje Curie (supervivencia sin complicaciones [SSC] a 5 años, 43,0 ± 5,7 % [puntaje Curie ≤12] vs. 21,4 ± 3,6 % [puntaje Curie >12], P < 0,0001). El límite óptimo del puntaje Curie después de la quimioterapia de inducción fue de 2 en SIOPEN/HR-NBL1, un puntaje Curie posinducción superior a 2 se relacionó con un desenlace inferior (tasa de SSC a 5 años, 39,2 ± 4,7 % [puntaje Curie ≤2] vs. 16,4 ± 4,2 % [puntaje Curie >2], P < 0,0001). El puntaje Curie posinducción mantuvo la significación estadística independiente en los modelos de Cox cuando se ajustó según las covariables de edad y número de copias del gen MYCN.[88 ] - Método de puntaje SIOPEN: es un sistema de puntaje independiente formulado por SIOPEN para las gammagrafías con MIBG que, en comparación con el sistema de puntaje Curie, divide el cuerpo en 12 segmentos, en lugar de 10, y en cada segmento se asignan 6 grados, en lugar de 4 grados para la absorción de MIBG.[
99 ] Posteriormente, el sistema de puntaje SIOPEN se validó en forma independiente con datos de un segundo ensayo clínico grande.[1010 ]
El German Pediatric Oncology Group comparó el valor pronóstico de los métodos de puntajes Curie y SIOPEN en un estudio retrospectivo con 58 pacientes de neuroblastoma en estadio 4, mayores de 1 año de vida. Se observaron resultados muy similares. En el momento del diagnóstico, un puntaje Curie de 2 o inferior, y un puntaje SIOPEN de 4 o inferior (el mejor valor límite) se correlacionaron con tasas de SSC y supervivencia general (SG) significativamente mejores, en comparación con puntajes más altos. Al cabo de 4 ciclos de quimioterapia de inducción, los pacientes con respuesta completa según los puntajes SIOPEN y Curie tuvieron mejores desenlaces que los pacientes con captación residual en las metástasis; sin embargo, la resolución subsiguiente de las metástasis captadoras de MIBG entre los ciclos 4 y 6 de quimioterapia no afectó al pronóstico.[
Los ensayos clínicos mencionados no incluyeron evaluaciones de la fase de posinducción de los puntajes Curie o SIOPEN después del trasplante y la inmunoterapia; los valores límite y los desenlaces relacionados con estas evaluaciones quizás difieran de los que se obtienen con los puntajes de preinducción y posinducción.
Tomografía por emisión de positrones
Se utilizan tomografías por emisión de positrones (TEP) con flúor F 18-fluorodesoxiglucosa para evaluar la extensión de la enfermedad en pacientes con tumores que no son ávidos de MIBG.[
Otras pruebas y procedimientos de estadificación
Otras pruebas y procedimientos que se utilizan para la estadificación del neuroblastoma son los siguientes.
- Aspiración de la médula ósea y biopsia: la médula ósea se evalúa mediante aspiración bilateral en la cresta ilíaca y biopsias con trépano (por punción con aguja gruesa) para excluir el compromiso de la médula ósea. Para que las muestras de la biopsia por punción con aguja gruesa se consideren adecuadas deben contener, por lo menos, 1 cm de médula, sin cartílago. En muchos estudios del COG se exigen dos biopsias por punción con aguja gruesa y dos aspiraciones. Las muestras de la médula ósea tal vez sean innecesarias para los tumores que, de otra forma, están en estadio 1.[
1212 ] - Evaluación de ganglios linfáticos: cuando se usa la estadificación del INSS para evaluar la extensión de la enfermedad, se hace una evaluación clínica y confirmación patológica del compromiso de los ganglios linfáticos palpables.[
11 ] Se usan una TC, imágenes por resonancia magnética (IRM), o ambos tipos de imágenes para evaluar ganglios linfáticos en regiones que no se pueden identificar con facilidad durante el examen físico. El sistema de estadificación del International Neuroblastoma Risk Group (INRG) no exige la evaluación de los ganglios linfáticos, si bien la presencia de masas ganglionares incide en los factores de riesgo definidos por imágenes (IDRF). Para obtener más información, consultar las listas de los IDRF (IDRF originales IDRF originales eIDRF del COGIDRF del COG ). - Tomografía computada e imágenes por resonancia magnética:
- Se obtienen imágenes tridimensionales del tumor primario y los sitios posibles de drenaje linfático mediante TC o IRM del tórax, el abdomen y la pelvis. En general, las ecografías se consideran subóptimas para mediciones tridimensionales exactas.
- Es posible que los tumores paraespinales se diseminen a través de los agujeros intervertebrales y compriman la médula espinal. Por lo tanto, la IRM de la sección de la columna adyacente a cualquier tumor paraespinal es parte de la evaluación para la estadificación.
- Se realizan una TC o IRM de encéfalo y órbita si hay una indicación clínica según el examen médico o la gammagrafía con MlBG.
- En un estudio de 50 niños con neuroblastoma (todos ellos con tumores primarios en el abdomen o la pelvis) se evaluó la función del contraste de gadolinio como parte de las exploraciones con IRM. La evaluación del tamaño del tumor y de los IDRF resultó similar, con independencia de si se usó o no gadolinio.[
1313 ]
Se evita la punción lumbar porque las metástasis en el sistema nervioso central (SNC) son infrecuentes en el momento del diagnóstico;[
Sistemas de estadificación del neuroblastoma
Durante décadas se han usado combinaciones de factores pronósticos (características clínicas y biológicas) para estratificar el riesgo de los pacientes e informar sobre la asignación del tratamiento.[
International Neuroblastoma Staging System
El International Neuroblastoma Staging System (INSS) se creó y adoptó por el COG en 1986 y por grupos cooperativos de Europa y Japón en 1993. El INSS es un sistema de estadificación posquirúrgica en el que se usa la localización del tumor con respecto a las estructuras de la línea media, el estado ganglionar y, lo que es más importante, la extensión de la resección quirúrgica inicial con el fin de determinar si un tumor locorregional se encuentra en estadio 1, 2A, 2B o 3 del INSS.[
International Neuroblastoma Risk Group Staging System
Con el propósito de elaborar un sistema de estadificación independiente de la extensión de la resección quirúrgica, el INRGSS se creó en 2005 mediante el uso de factores de riesgo definidos por imagen (IDRF) para categorizar los tumores locorregionales como L1 (IDRF ausentes), L2 (IDRF presentes), M (metastásicos) o MS (el equivalente a 4S del INSS). En numerosos estudios, la presencia de IDRF se ha relacionado con un aumento de complicaciones intraoperatorias, resección tumoral incompleta y peor supervivencia.[
| Estadio | Descripción |
|---|---|
| IDRF = factores de riesgo definidos por imágenes; INSS = International Neuroblastoma Staging System. | |
| a Adaptado de Monclair et al.[ |
|
| L1 | Tumor localizado sin compromiso de estructuras vitales según la definición de la lista de IDRF,a y limitado a un compartimiento específico del cuerpo. |
| L2 | Tumor locorregional con presencia de uno o más IDRF.a |
| M | Metástasis a distancia (excepto estadio MS). |
| MS | Metástasis limitadas a la piel, el hígado o la médula ósea en niños menores de 18 meses. El tumor primario puede estar en estadio 1, 2 o 3 del INSS. |
Conforme se definen en la bibliografía original, los IDRF comprenden los siguientes aspectos:[
- Extensión tumoral ipsilateral a dos compartimientos corporales: cuello y tórax; tórax y abdomen; o abdomen y pelvis.
- Infiltración de órganos o estructuras adyacentes: pericardio, diafragma, riñón, hígado, bloque duodenopancreático o mesenterio.
- Encapsulamiento tumoral de vasos sanguíneos mayores: arteria vertebral, yugular interna, vasos subclavios, arteria carótida, aorta, vena cava, vasos torácicos mayores, ramas en la raíz de la arteria mesentérica superior y el tronco celíaco, y vasos ilíacos.
- Compresión de la tráquea o los bronquios principales.
- Encapsulamiento del plexo braquial.
- Infiltración a nivel portohepático o del ligamento hepatoduodenal.
- Infiltración de la unión costovertebral entre T9 y T12.
- Tumor que cruza la escotadura ciática.
- Tumor que invade el pedículo renal.
- Extensión del tumor a la base del cráneo.
- Extensión tumoral intraespinal con invasión de más de un tercio del canal espinal, desaparición del espacio leptomeníngeo o señal anómala de la médula espinal en la IRM.
Los IDRF del COG, en los que se usa un abordaje de localización anatómica, son los siguientes:[
- Nuca y unión cervicotorácica: compromiso o encapsulamiento tumoral del plexo braquial, los vasos subclavios o la arteria carótida o vertebral, la vena yugular interna, la base del cráneo; compresión tumoral de la tráquea.
- Tórax: compromiso o encapsulamiento tumoral de la aorta o sus ramas principales; compresión tumoral de la tráquea o los bronquios principales; tumor en mediastino inferior, infiltración de la unión costovertebral entre T9 y T12
- Toracoabdominal: compromiso o encapsulamiento tumoral de la aorta o la vena cava.
- Abdomen y pelvis: compromiso o encapsulamiento tumoral del hilio hepático o el ligamento hepatoduodenal, la arteria mesentérica superior en la raíz, el origen del tronco celíaco o de la arteria mesentérica; compromiso o encapsulamiento tumoral de uno o ambos pedículos renales, la aorta o la vena cava; encapsulamiento o compromiso tumoral de los vasos ilíacos; tumor pélvico con compromiso o encapsulamiento de la escotadura ciática.
- Diseminación intraespinal del tumor: invade más de un tercio del plano transversal o los espacios leptomeníngeos perimedulares no son visibles; señal anómala de la médula espinal; tumores con forma de mancuerna con síntomas de compresión de la médula espinal.
- Compromiso o infiltración de órganos o estructuras adyacentes en cualquier localización: pericardio, diafragma, riñón, hígado, bloque duodenopancreático, mesenterio y otros.
- Tumor que compromete dos compartimientos corporales: cuello y tórax; tórax y abdomen; abdomen y pelvis.
- Sin IDRF, pero con afecciones registradas: derrame pleural multifocal, ascitis.
La evaluación de la resecabilidad quirúrgica debe incluir los IDRF. A medida que se incrementa el número de IDRF, aumenta la morbilidad quirúrgica y disminuye la posibilidad de resección completa.
La quimioterapia neoadyuvante no siempre elimina de manera eficaz los IDRF, conforme se observó en el ensayo retrospectivo European Unresectable Neuroblastoma realizado entre 2001 y 2006, en el que se analizaron datos de 143 pacientes mayores de 1 año con neuroblastoma en estadio 3 del INSS sin amplificación de MYCN. Todos los pacientes tenían factores de riesgo quirúrgico que indicaban la irresecabilidad de los tumores. En una revisión central de un subconjunto se encontraron características histológicas desfavorables, según la International Neuroblastoma Pathology Classification, en el 53 % de los pacientes. En el momento del diagnóstico, se identificaron 228 IDRF.[
- Después de 4 ciclos de quimioterapia con carboplatino y etopósido alternados con vincristina, ciclofosfamida y doxorrubicina, solo el 32,2 % de los pacientes presentaron resolución de los IDRF, el 49 % de los pacientes no presentaron cambios en los IDRF y el 18,8 % de los pacientes presentaron IDRF nuevos.
- La resección completa fue posible en el 71,2 % de los pacientes en quienes los IDRF disminuyeron o desaparecieron. Se logró la resección completa o casi completa, en el 84 % de los pacientes (37 de 44) en quienes los IDRF disminuyeron o desaparecieron. Se logró la resección completa o casi completa, en el 70 % de los pacientes (39 de 56) con IDRF estables y en el 52 % de los pacientes (13 de 25) con IDRF nuevos.
- No se observaron diferencias significativas en la SSC o la SG conforme a la respuesta a la quimioterapia de los IDRF y los resultados quirúrgicos. No se estableció ninguna asociación entre el tipo de IDRF antes de la cirugía y el alcance de la resección.
- Cuando el tumor rodeaba la arteria mesentérica superior o el tronco celíaco, se vieron afectadas la supervivencia sin enfermedad (SSE) y la SG (tal vez por la dificultad para lograr una resección completa en estas áreas).
- La quimioterapia prolongada con más de 5 cursos no ayudó a reducir los IDRF y se relacionó con SSE y SG más bajas.
El INRGSS incorporó este sistema de estadificación en un sistema de grupos del riesgo mediante la aplicación de múltiples parámetros en el momento del diagnóstico.[
El INRGSS simplifica los estadios y usa L1, L2, M o MS. Los tumores localizados se clasifican como enfermedad en estadio L1 o L2, según la presencia de 1 o más de los 20 IDRF.[
En la colaboración con el INRG también se definieron técnicas para detectar y cuantificar el neuroblastoma en la médula ósea, tanto en el momento del diagnóstico como después del tratamiento. La cuantificación de la enfermedad metastásica en la médula ósea podría dar lugar a evaluaciones más exactas de la respuesta al tratamiento,[
La decisión del INRG Task Force de reemplazar la categoría de enfermedad 4S con la de la nueva definición de MS se tomó a partir de los informes de desenlaces favorables en un número pequeño de lactantes con tumores primarios L2 y patrones metastásicos, incluso aquellos de 12 a 18 meses de vida.[
Al combinar el INRGSS, la edad y los factores biológicos, se asigna a cada paciente a un grupo de riesgo del INRG que predice el desenlace y orienta en cuanto al abordaje adecuado de tratamiento según el riesgo. La validez del INRGSS se analizó en los siguientes estudios retrospectivos de neuroblastoma localizado sin amplificación de MYCN y con estadio del INRGSS definido antes:
- En el primer estudio se analizaron datos de un ensayo de la SIOPEN y se encontraron tumores L2 en pacientes en estadio 1 (21 %), estadio 2 (45 %) y estadio 3 (94 %) del INSS. El INRGSS tuvo un valor predictivo para los desenlaces: los pacientes en estadio L1 tuvieron una tasa de SSC a 5 años del 90 % y una tasa de SG del 96 %, versus una tasa de SSC del 79 % y una tasa de SG del 89 % para los pacientes en estadio L2.[
2121 ] - En el segundo estudio se usaron datos del ensayo multicéntrico europeo LNESG1, sobre cirugía inicial seguida por observación, que se realizó entre 1995 y 1999; 291 niños tuvieron tumores L1 y todos se sometieron a cirugía inicial. De los pacientes en estadio L2, 118 se sometieron a cirugía inicial y 125 no se sometieron a cirugía alguna (106 niños del segundo grupo recibieron quimioterapia neoadyuvante).[
2929 ]- Las tasas de SSC y de SG a 5 años fueron del 92 % y el 98 % para el grupo L1; del 86 % y el 95 % para el grupo L2 con cirugía inicial; y del 73 % y el 83 % para el grupo L2 sin cirugía inicial.
- Cabe destacar que muchos niños con tumores L2 se sometieron a cirugía inicial y tuvieron un desenlace significativamente superior al de los niños sometidos a biopsia sola como el procedimiento quirúrgico inicial (tasa de SG a 5 años del 93 vs. 83 %). Es posible que los tumores L2 que se resecaron de manera primaria se seleccionaran debido a una resecabilidad de menor riesgo. Sin embargo, estos niños también tuvieron una tasa de complicaciones quirúrgicas del 17 % (vs. el 5 % para las resecciones del grupo L1).
- Entre los pacientes sometidos a cirugía inicial, aquellos que presentaron complicaciones quirúrgicas tuvieron una tasa de SG más baja (92 vs. 97 %, P = 0,05), pero este efecto en el desenlace fue estadísticamente significativo solo en pacientes con tumores L1.
- Para los pacientes del grupo L2, las complicaciones quirúrgicas no guardaron relación estadística con los IDRF.[
2929 ]
En la mayoría de los protocolos internacionales se están empezando a incorporar la recopilación y uso de los IDRF para definir el estadio INRG, que se usa en la estratificación del riesgo y la asignación del tratamiento.[
Agrupamiento por riesgo para el neuroblastoma del Children's Oncology Group
El estudio biológico del COG ANBL00B1 (NCT00904241) sirve de infraestructura para la adquisición rápida y fiable de marcadores de pronóstico tumoral usados para la clasificación del riesgo y la elegibilidad para ensayos clínicos. Para obtener más información sobre las categorías de riesgo del COG, consultar el
Agrupamiento por riesgo del International Neuroblastoma Risk Group
En el esquema de clasificación del International Neuroblastoma Risk Group (INRG), se asigna a los pacientes de neuroblastoma a 1 de 16 grupos de riesgo pretratamiento a partir del estadio del INRG, la edad, la categoría histológica, el grado de diferenciación tumoral, la amplificación de MYCN, la anomalía en 11q (la única anomalía cromosómica segmentaria estudiada) y la ploidía. Se definieron 4 niveles de riesgo de acuerdo con los desenlaces de 8800 pacientes de los que se obtuvieron datos de alta calidad a medida que ingresaron en ensayos clínicos (consultar el
El COG analizó a casi 5000 pacientes con neuroblastoma recién diagnosticado que se inscribieron en el estudio ANBL00B1 entre 2007 y 2017 y que recibieron tratamientos más modernos. Además de utilizar el INRGSS, se examinó el estado cromosómico segmentario de 1q con los factores pronósticos anteriores.[
En el agrupamiento general por riesgo, el tipo histológico es un determinante importante del riesgo para todos los tumores en estadios L1 y L2; el grado de diferenciación separa a los neuroblastomas de los ganglioneuroblastomas nodulares en pacientes mayores de 18 meses. Las metas del INRG son aumentar la colaboración internacional y clasificar a los pacientes de manera uniforme con el fin de comparar los resultados de los ensayos clínicos que se realizan en todo el mundo.[
| Estadio del INRG | Categoría histológica | Grado de diferenciación tumoral | MYCN | Anomalía en 11q | Ploidía | Grupo de riesgo pretratamiento | |
|---|---|---|---|---|---|---|---|
| GN = ganglioneuroma; GNB = ganglioneuroblastoma. | |||||||
| a Reproducción autorizada. © (2015) American Society of Clinical Oncology. Todos los derechos reservados. Pinto N et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma, J Clin Oncol 33 (27), 2015: 3008–3017.[ |
|||||||
| L1/L2 | GN en maduración, GNB entremezclado | A (muy bajo) | |||||
| L1 | Cualquiera, excepto GN en maduración o GNB entremezclado | No amplificado | B (muy bajo) | ||||
| Amplificado | K (alto) | ||||||
| L2 | |||||||
| Edad <18 meses | Cualquiera, excepto GN en maduración o GNB entremezclado | No amplificado | No | D (bajo) | |||
| Sí | G (intermedio) | ||||||
| Edad ≥18 meses | GNB, neuroblastoma nodular | En diferenciación | No amplificado | No | E (bajo) | ||
| Sí | H (intermedio) | ||||||
| Poco diferenciado o indiferenciado | No amplificado | H (intermedio) | |||||
| Amplificado | N (alto) | ||||||
| M | |||||||
| Edad <18 meses | No amplificado | Hiperdiploidía | F (bajo) | ||||
| Edad <12 meses | No amplificado | Diploidía | I (intermedio) | ||||
| Edad 12 a <18 meses | No amplificado | Diploidía | J (intermedio) | ||||
| Edad <18 meses | No amplificado | O (alto) | |||||
| Edad ≥18 meses | P (alto) | ||||||
| MS | |||||||
| Edad <18 meses | No amplificado | No | C (muy bajo) | ||||
| Sí | Q (alto) | ||||||
| Amplificado | R (alto) | ||||||
Referencias:
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.
- Howman-Giles R, Shaw PJ, Uren RF, et al.: Neuroblastoma and other neuroendocrine tumors. Semin Nucl Med 37 (4): 286-302, 2007.
- Papathanasiou ND, Gaze MN, Sullivan K, et al.: 18F-FDG PET/CT and 123I-metaiodobenzylguanidine imaging in high-risk neuroblastoma: diagnostic comparison and survival analysis. J Nucl Med 52 (4): 519-25, 2011.
- Gauguet JM, Pace-Emerson T, Grant FD, et al.: Evaluation of the utility of (99m) Tc-MDP bone scintigraphy versus MIBG scintigraphy and cross-sectional imaging for staging patients with neuroblastoma. Pediatr Blood Cancer 64 (11): , 2017.
- Kushner BH, Kramer K, Modak S, et al.: Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. J Clin Oncol 27 (7): 1041-6, 2009.
- Sharp SE, Shulkin BL, Gelfand MJ, et al.: 123I-MIBG scintigraphy and 18F-FDG PET in neuroblastoma. J Nucl Med 50 (8): 1237-43, 2009.
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.
- Yanik GA, Parisi MT, Naranjo A, et al.: Validation of Postinduction Curie Scores in High-Risk Neuroblastoma: A Children's Oncology Group and SIOPEN Group Report on SIOPEN/HR-NBL1. J Nucl Med 59 (3): 502-508, 2018.
- Lewington V, Lambert B, Poetschger U, et al.: 123I-mIBG scintigraphy in neuroblastoma: development of a SIOPEN semi-quantitative reporting ,method by an international panel. Eur J Nucl Med Mol Imaging 44 (2): 234-241, 2017.
- Ladenstein R, Lambert B, Pötschger U, et al.: Validation of the mIBG skeletal SIOPEN scoring method in two independent high-risk neuroblastoma populations: the SIOPEN/HR-NBL1 and COG-A3973 trials. Eur J Nucl Med Mol Imaging 45 (2): 292-305, 2018.
- Decarolis B, Schneider C, Hero B, et al.: Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol 31 (7): 944-51, 2013.
- Russell HV, Golding LA, Suell MN, et al.: The role of bone marrow evaluation in the staging of patients with otherwise localized, low-risk neuroblastoma. Pediatr Blood Cancer 45 (7): 916-9, 2005.
- Morin CE, Hasweh R, Anton C, et al.: Gadolinium-based contrast media does not improve the staging of neuroblastoma image-defined risk factors at diagnosis. Pediatr Blood Cancer 71 (1): e30724, 2024.
- DuBois SG, Kalika Y, Lukens JN, et al.: Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol 21 (3): 181-9, 1999 May-Jun.
- Kramer K, Kushner B, Heller G, et al.: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91 (8): 1510-9, 2001.
- Pinto NR, Applebaum MA, Volchenboum SL, et al.: Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J Clin Oncol 33 (27): 3008-17, 2015.
- Cohn SL, Pearson AD, London WB, et al.: The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27 (2): 289-97, 2009.
- Brodeur GM, Seeger RC, Barrett A, et al.: International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6 (12): 1874-81, 1988.
- Castleberry RP, Shuster JJ, Smith EI: The Pediatric Oncology Group experience with the international staging system criteria for neuroblastoma. Member Institutions of the Pediatric Oncology Group. J Clin Oncol 12 (11): 2378-81, 1994.
- Ikeda H, Iehara T, Tsuchida Y, et al.: Experience with International Neuroblastoma Staging System and Pathology Classification. Br J Cancer 86 (7): 1110-6, 2002.
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.
- Brisse HJ, McCarville MB, Granata C, et al.: Guidelines for imaging and staging of neuroblastic tumors: consensus report from the International Neuroblastoma Risk Group Project. Radiology 261 (1): 243-57, 2011.
- Newman EA, Nuchtern JG: Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg 25 (5): 257-264, 2016.
- Chen AM, Trout AT, Towbin AJ: A review of neuroblastoma image-defined risk factors on magnetic resonance imaging. Pediatr Radiol 48 (9): 1337-1347, 2018.
- Avanzini S, Pio L, Erminio G, et al.: Image-defined risk factors in unresectable neuroblastoma: SIOPEN study on incidence, chemotherapy-induced variation, and impact on surgical outcomes. Pediatr Blood Cancer 64 (11): , 2017.
- Burchill SA, Beiske K, Shimada H, et al.: Recommendations for the standardization of bone marrow disease assessment and reporting in children with neuroblastoma on behalf of the International Neuroblastoma Response Criteria Bone Marrow Working Group. Cancer 123 (7): 1095-1105, 2017.
- Park JR, Bagatell R, Cohn SL, et al.: Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 35 (22): 2580-2587, 2017.
- Taggart DR, London WB, Schmidt ML, et al.: Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol 29 (33): 4358-64, 2011.
- Monclair T, Mosseri V, Cecchetto G, et al.: Influence of image-defined risk factors on the outcome of patients with localised neuroblastoma. A report from the LNESG1 study of the European International Society of Paediatric Oncology Neuroblastoma Group. Pediatr Blood Cancer 62 (9): 1536-42, 2015.
- Cecchetto G, Mosseri V, De Bernardi B, et al.: Surgical risk factors in primary surgery for localized neuroblastoma: the LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J Clin Oncol 23 (33): 8483-9, 2005.
- Simon T, Hero B, Benz-Bohm G, et al.: Review of image defined risk factors in localized neuroblastoma patients: Results of the GPOH NB97 trial. Pediatr Blood Cancer 50 (5): 965-9, 2008.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es infrecuente, aunque desde 1975 se ha observado un aumento gradual de la incidencia general.[
- Médico de atención primaria.
- Patólogos pediatras.
- Cirujanos pediatras.
- Radioncólogos pediatras.
- Oncólogos o hematólogos pediatras.
- Enfermeros especializados en pediatría.
- Trabajadores sociales.
- Profesionales de la vida infantil.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes de cáncer infantil.[
Referencias:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014.
Also available onlineAlso available online . Last accessed August 23, 2024.
Aspectos generales de las opciones de tratamiento del neuroblastoma
El tratamiento se basa en el riesgo. En el sistema de determinación del riesgo del Children's Oncology Group (COG), cada niño se asigna a un grupo de riesgo bajo, riesgo intermedio o riesgo alto a partir de los siguientes factores:[
- Estadio del International Neuroblastoma Staging System (INSS).
- Edad.
- International Neuroblastoma Pathologic Classification (INPC).[
88 ] - Ploidía.
- Amplificación del oncogén MYCN en el tejido tumoral.[
22 ,33 ,44 ,55 ,66 ,77 ]
La evaluación del riesgo para el neuroblastoma en estadio bajo con amplificación de MYCN es objeto de polémica por su escasa frecuencia.
En un estudio con 87 pacientes de neuroblastoma en estadio 1 y estadio 2 del INSS con amplificación de MYCN, tomados de varios grupos de ensayos clínicos, se mostró que la edad, el estadio y el tratamiento inicial no tuvieron efecto en el desenlace. La tasa de supervivencia sin complicaciones (SSC) fue del 53 % y la tasa de supervivencia general (SG) fue del 72 %. La supervivencia fue superior en pacientes cuyos tumores eran hiperdiploides en vez de diploides (tasa de SSC, 82 ± 20 % vs. 37 ± 21 %; tasa de SG, 94 ± 11 % vs. 54 ± 15 %).[
El COG considera que los lactantes con enfermedad en estadio 4 y 4S, y con amplificación de MYCN tienen riesgo alto.[
Para obtener más información sobre las categorías de riesgo del COG, consultar el
Otros factores biológicos que influyeron la selección del tratamiento en algunos estudios del COG previos fueron la pérdida desequilibrada de heterocigosis en 11q y la pérdida de heterocigosidad del cromosoma 1p.[
| Riesgo | Estadio | Edad | Estado deMYCN | Ploidía del DNA | INPC | Otras |
|---|---|---|---|---|---|---|
| ID = índice de DNA; HF = tipo histológico favorable; INPC = International Neuroblastoma Pathology Classification; HD = tipo histológico desfavorable. | ||||||
| a Adaptado de Liang et al.[ |
||||||
| b Grupo de riesgo bajo, según se define en el ensayo del Children's Oncology GroupANBL00B1 (NCT00904241). | ||||||
| c Grupo de riesgo intermedio, según se define en el ensayo del Children's Oncology GroupANBL0531 (NCT00499616). | ||||||
| d Grupo de riesgo alto, según se define en el ensayo del Children's Oncology GroupANBL0532 (NCT00567567). | ||||||
| Bajo b | 1 | Cualquiera | Cualquiera | Cualquiera | Cualquiera | |
| 2a/2b | Cualquiera | No amplificado | Cualquiera | Cualquiera | Resección ≥50 % | |
| 4s | <12 meses | No amplificado | ID >1 | HF | Asintomático | |
| Intermedio c | 2a/2b | 0–12 años | No amplificado | Cualquiera | Cualquiera | Biopsia o resección <50 % |
| 3 | <18 meses | No amplificado | Cualquiera | Cualquiera | ||
| 3 | ≥18 meses a 12 años | No amplificado | Cualquiera | HF | ||
| 4 | <12 meses | No amplificado | Cualquiera | Cualquiera | ||
| 4 | 12 meses a <18 meses | No amplificado | ID >1 | HF | ||
| 4s | <12 meses | No amplificado | Cualquiera | Cualquiera | Sintomático | |
| 4s | <12 meses | No amplificado | ID = 1 | Cualquiera | Asintomático o sintomático | |
| 4s | <12 meses | No amplificado | Cualquiera | HD | Asintomático o sintomático | |
| 4s | <12 meses | Faltante | Faltante | Faltante | Demasiado enfermo para someterse a biopsia | |
| Alto d | 2a/2b | Cualquiera | Amplificado | Cualquiera | Cualquiera | Cualquier grado de resección |
| 3 | Cualquiera | Amplificado | Cualquiera | Cualquiera | ||
| 3 | ≥18 meses | No amplificado | Cualquiera | HD | ||
| 4 | <12 meses | Amplificado | Cualquiera | Cualquiera | ||
| 4 | 12 meses a <18 meses | Amplificado | Cualquiera | Cualquiera | ||
| 4 | 12 meses a <18 meses | Cualquiera | ID = 1 | Cualquiera | ||
| 4 | 12 meses a <18 meses | Cualquiera | Cualquiera | HD | ||
| 4 | ≥18 meses | Cualquiera | Cualquiera | Cualquiera | ||
| 4s | <12 meses | Amplificado | Cualquiera | Cualquiera | Asintomático o sintomático | |
Por lo general, el tratamiento se basa en la clasificación del tumor en riesgo bajo, intermedio o alto, conforme consta a continuación:
- Riesgo bajo. Para los pacientes con tumores de riesgo bajo, el abordaje es la observación o la resección, y quimioterapia solo para los pacientes sintomáticos con características biológicas de riesgo bajo. La tasa de SG a 5 años fue del 98 % para los pacientes de riesgo bajo entre los más de 5000 pacientes inscritos en el estudio biológico del COG ANBL00B1 (NCT00904241).[
1414 ] En el estudio del COG en curso se está analizando la reducción del tratamiento en un subconjunto limitado de pacientes con tumores de riesgo bajo. - Riesgo intermedio. Para pacientes con tumores de riesgo intermedio, es frecuente la administración de quimioterapia antes de la resección definitiva y se usa quimioterapia multifarmacológica que incluye doxorrubicina, ciclofosfamida, un fármaco derivado del platino y etopósido. El número de ciclos de quimioterapia depende de los factores de riesgo clínicos y biológicos del tumor, así como de la respuesta al tratamiento.[
1313 ] El objetivo de la quimioterapia es administrar quimioterapia con una duración suficiente (con cirugía o sin esta) para lograr, como mínimo, una respuesta parcial (reducción de al menos el 50 % de las masas de tejido blando) y la resolución de la enfermedad metastásica.[1313 ] En estudios recientes, se observó a determinados pacientes sin someterlos a quimioterapia ni intentar una resección. La tasa de SG a 5 años fue de alrededor del 95 % para los pacientes de riesgo intermedio entre los más de 5000 pacientes inscritos en el estudio biológico del COG ANBL00B1 (NCT00904241).[1414 ] En el estudio del COG ANBL0531 (NCT00499616) , la duración y la intensidad de la quimioterapia se redujo en varios subconjuntos de niños de riesgo intermedio para reducir aún más los efectos secundarios; no se observó empeoramiento de los desenlaces.[1313 ] - Riesgo alto. Para los pacientes de riesgo alto, el tratamiento se intensifica con quimioterapia, cirugía, radioterapia, terapia mielosupresora y trasplante de células madre hematopoyéticas (TCMH), isotretinoína e inmunoterapia, lo que produce tasas de supervivencia a 5 años del 62 %.[
1414 ] En un estudio aleatorizado de fase III del COG (ANBL0532 [NCT00567567]) se observó una mejora estadísticamente significativa de la supervivencia con ciclos en tándem de terapia mielosupresora y TCMH, en comparación con un ciclo único de terapia mielosupresora y TCMH. La tasa de SSC a 3 años en los pacientes que recibieron trasplantes en tándem fue superior (P = 0,006) a la tasa de SSC en los pacientes que recibieron trasplantes únicos; sin embargo, hubo un posible sesgo de selección como resultado de una gran proporción de pacientes que no se aleatorizaron.[1515 ][Nivel de evidencia A1] Para obtener más información, consultar la secciónFase de consolidaciónFase de consolidación .
En el
| Asignación al grupo de riesgo del COG | Opciones de tratamiento | |
|---|---|---|
| COG = Children's Oncology Group; GM-CSF = factor estimulante de colonias de granulocitos y macrófagos; 131I-MlBG = yodo I 131-metayodobencilguanidina; TCMH = trasplante de células madre hematopoyéticas. | ||
| Neuroblastoma de riesgo bajo | |
|
| |
||
| |
||
| |
||
| Neuroblastoma de riesgo intermedio | |
|
| |
||
| |
||
| Neuroblastoma de riesgo alto | |
|
| Neuroblastoma en estadio 4S o MS | |
|
| |
||
| |
||
International Neuroblastoma Response Criteria revisados
Los International Neuroblastoma Response Criteria (INRC) se usan para evaluar la respuesta al tratamiento.[
Los criterios de respuesta generales de los INRC se definen de la siguiente manera:[
- Respuesta completa: no hay indicios de enfermedad, incluye la resolución de la captación de MIBG (o del resultado positivo de la TEP para la enfermedad sin avidez de MIBG) en cualquier sitio de tejido blando o hueso y el compromiso residual mide menos de 10 mm en una imagen tridimensional del tumor primario; los ganglios linfáticos diana miden menos de 15 mm en su dimensión corta y no hay indicios histológicos de tumor en 2 biopsias de médula ósea y 2 muestras de aspiraciones de médula ósea en un punto de referencia.
- Respuesta parcial: disminución del 30 % o más del diámetro mayor del sitio primario y ausencia de lesiones nuevas y estabilidad o mejora de la captación de MIBG (o TEP con 18F-FDG) y una reducción de, por lo menos, un 50 % en el puntaje óseo total de MIBG o una reducción de un 50 % o mayor en el número de lesiones óseas ávidas en la TEP con 18F-FDG.
- Respuesta menor: respuesta parcial o respuesta completa de, al menos, un componente de la enfermedad, pero hay como mínimo un componente de enfermedad estable y ningún componente de enfermedad progresiva.
- Enfermedad progresiva: cualquier lesión nueva; un aumento de un 20 % en el diámetro mayor de cualquier lesión medible y aumento de, por lo menos, 5 mm en el diámetro mayor; la médula ósea que no tenía compromiso inicial, ahora muestra compromiso tumoral; cualquier lesión nueva en el tejido blando que es ávida de MIBG (o TEP con 18F-FDG) o que se confirma su compromiso mediante biopsia; un nuevo sitio óseo ávido; o aumento de un 1,25 % o más del puntaje relativo de la MIBG.
- Enfermedad estable: reducción insuficiente para definir una respuesta parcial o aumento insuficiente para definir una enfermedad progresiva, y es posible encontrar infiltración tumoral mayor al 5 % como se define en la enfermedad mínima.
Se debe interpretar con cuidado la evolución de la enfermedad metastásica en un lactante clasificado al inicio con enfermedad en estadio 1 o 2. Si el patrón de las metástasis en dicho paciente es congruente con un patrón de enfermedad 4S (compromiso de piel, hígado, o de menos del 10 % de médula ósea), estos pacientes no se incluyen en la categoría de enfermedad progresiva o metastásica, que sería un criterio típico para excluirlos del protocolo de tratamiento. Más bien, estos pacientes se tratan como los pacientes en estadio 4S.
Hay polémica en torno a la necesidad de usar una medición tridimensional del tumor primario para definir la respuesta o si esto es igual a medir la dimensión más larga, como se hace para determinar la respuesta tumoral según RECIST.[
Cirugía
En pacientes sin enfermedad metastásica, el estándar de atención es someterlos a una cirugía inicial. El objetivo de dicha cirugía, sobre la base del estadio de la enfermedad y el grupo de riesgo, es lograr los siguientes objetivos:
- Obtener tejido para el diagnóstico. La biopsia incisional o la biopsia con aguja gruesa solo se recomiendan para pacientes con enfermedad en estadio L2,[
2020 ][Nivel de evidencia C1] y por lo general no debe intentarse una resección inicial. Para obtener más información sobre los factores de riesgo definidos por imágenes, consultar la secciónInternational Neuroblastoma Risk Group Staging System (INRGSS)International Neuroblastoma Risk Group Staging System (INRGSS) .- Tanto la biopsia incisional como la biopsia percutánea con aguja gruesa son aceptables para los pacientes con enfermedad en estadio L2. En un estudio retrospectivo multiinstitucional no se observó diferencias significativas en la capacidad de obtener con precisión un diagnóstico primario mediante biopsia percutánea con aguja gruesa, en comparación con la biopsia incisional (95,7 vs. 98,9 %, P = 0,314), o determinar el número de copias de MYCN (92,4 vs. 97,8 %; P = 0,111). El rendimiento de la pérdida de heterocigosidad y la ploidía tumoral fue menor con la biopsia percutánea con aguja gruesa (en función del número de núcleos o volumen de tejido obtenido) que con la biopsia incisional (56,1 vs. 90,9 %, P < 0,05; y 58,0 vs. 88,5 %, P < 0,05). Las complicaciones no difirieron entre los grupos.[
2121 ][Nivel de evidencia C1]
- Tanto la biopsia incisional como la biopsia percutánea con aguja gruesa son aceptables para los pacientes con enfermedad en estadio L2. En un estudio retrospectivo multiinstitucional no se observó diferencias significativas en la capacidad de obtener con precisión un diagnóstico primario mediante biopsia percutánea con aguja gruesa, en comparación con la biopsia incisional (95,7 vs. 98,9 %, P = 0,314), o determinar el número de copias de MYCN (92,4 vs. 97,8 %; P = 0,111). El rendimiento de la pérdida de heterocigosidad y la ploidía tumoral fue menor con la biopsia percutánea con aguja gruesa (en función del número de núcleos o volumen de tejido obtenido) que con la biopsia incisional (56,1 vs. 90,9 %, P < 0,05; y 58,0 vs. 88,5 %, P < 0,05). Las complicaciones no difirieron entre los grupos.[
- Se recomienda la resección casi total o total del tumor primario en función del estadio.
- Esta es la norma estándar para los pacientes con enfermedad de riesgo bajo (excluidos los lactantes con diagnóstico prenatal que son candidatos a observación) y de riesgo intermedio. En pacientes con tumores L1 (los que no tienen factores de riesgo quirúrgico definidos por imágenes), los tumores son resecables con riesgo bajo de nefrectomía o complicaciones potencialmente mortales. Los tumores primarios unilaterales de la glándula suprarrenal, la enfermedad torácica en estadio L1 o la enfermedad del cuello en estadio L1 deben resecarse al inicio (a discreción del cirujano).[
2020 ][Nivel de evidencia C1] - Es posible considerar la cirugía mínimamente invasiva en determinados pacientes con neuroblastoma si la realiza un cirujano oncólogo pediátrico con experiencia en el uso de dicha técnica.[
2222 ][Nivel de evidencia C1]; [2323 ,2424 ]
- Esta es la norma estándar para los pacientes con enfermedad de riesgo bajo (excluidos los lactantes con diagnóstico prenatal que son candidatos a observación) y de riesgo intermedio. En pacientes con tumores L1 (los que no tienen factores de riesgo quirúrgico definidos por imágenes), los tumores son resecables con riesgo bajo de nefrectomía o complicaciones potencialmente mortales. Los tumores primarios unilaterales de la glándula suprarrenal, la enfermedad torácica en estadio L1 o la enfermedad del cuello en estadio L1 deben resecarse al inicio (a discreción del cirujano).[
El COG notificó que una conducta expectante de observación en lactantes menores de 6 meses con masas suprarrenales pequeñas (L1) produjo tasas de SSC y SG excelentes, al mismo tiempo que evitó una intervención quirúrgica en una gran mayoría de los pacientes.[
Continúa siendo polémico el hecho de si hay alguna ventaja en la resección macroscópica total de la masa del tumor primario después de la quimioterapia para pacientes en estadio 4 mayores de 18 meses.[
Para obtener más información sobre los factores de riesgo definidos por imágenes, consultar la sección
Radioterapia
En el paradigma de tratamiento vigente, la radioterapia para pacientes de neuroblastoma de riesgo bajo o intermedio, se reserva para masas tumorales sintomáticas potencialmente mortales o que ponen en peligro algún órgano que no respondió suficientemente rápido a la quimioterapia. Las situaciones comunes en las que se usa radioterapia para estos pacientes son las siguientes:
- Lactantes de 60 días de vida o menos en estadio 4S y compromiso respiratorio grave por metástasis hepáticas que no respondieron a la quimioterapia.[
3636 ] - Lactantes con compresión de la médula espinal. No obstante, la mayoría de los pacientes reciben tratamiento mediante quimioterapia o intervención neuroquirúrgica, debido al grado de respuesta del neuroblastoma a la quimioterapia y los posibles efectos tardíos graves de la radioterapia en niños pequeños.[
3737 ]
La radioterapia se ha convertido en parte del estándar de atención para los pacientes con enfermedad de riesgo alto y suele administrarse después de dosis altas de quimioterapia y del rescate de células madre. Para obtener más información, consultar la sección
Los datos de seguimiento a largo plazo del Childhood Cancer Survivor Study respaldan la limitación del uso de radioterapia en los lactantes con neuroblastoma (que en general no presentan enfermedad de riesgo alto). En este estudio se observaron tasas más elevadas de segundas neoplasias malignas y afecciones crónicas significativas en lactantes tratados con radioterapia.[
Tratamiento de la compresión de la médula espinal
La compresión de la médula espinal se considera una urgencia médica. Los pacientes se someten a tratamiento inmediato porque aumenta la probabilidad de recuperación neurológica cuando los síntomas están presentes por un período relativamente corto antes del diagnóstico y el tratamiento. La recuperación también depende de la gravedad de los defectos neurológicos (debilidad vs. parálisis). El resultado neurológico es similar, con independencia de si la compresión medular se trata con quimioterapia, radioterapia o cirugía, aunque la radioterapia se utiliza con menos frecuencia que en el pasado.
En los ensayos clínicos finalizados del COG sobre neuroblastomas, se recomendó la quimioterapia inmediata para la compresión medular en pacientes de riesgo bajo o intermedio.[
Es posible que una intervención neuroquirúrgica sea beneficiosa para los niños con compresión medular grave que no mejora de inmediato o cuyos síntomas empeoran. En algunas ocasiones, la laminectomía produce cifoescoliosis posterior y no elimina la necesidad de quimioterapia.[
La carga de problemas de salud a largo plazo en los supervivientes de neuroblastoma con diseminación intraespinal es alta. En una revisión sistemática de 28 estudios de tratamiento y resultado de pacientes con diseminación intraespinal, la gravedad de los síntomas en el momento del diagnóstico y las modalidades de tratamiento se asociaron más con la presencia de problemas de salud a largo plazo. En particular, la gravedad de los déficits motores neurológicos tuvo mayor probabilidad de predecir el desenlace neurológico.[
En una serie de 34 lactantes con compresión medular epidural sintomática, ni la cirugía ni la quimioterapia produjeron resultados satisfactorios una vez que se estableció la paraplejia. La frecuencia de los déficit motores de grado 3 y de la disfunción intestinal aumentó con un intervalo más prolongado de duración de los síntomas. La mayoría de los lactantes con compresión medular epidural sintomática presentaron secuelas, que fueron graves en alrededor de la mitad de los casos.[
Vigilancia durante y después del tratamiento
Aunque la función de las imágenes para la vigilancia en la detección de la recaída del neuroblastoma no se ha estudiado bien, la mayoría de los pacientes se someterán a pruebas con imágenes periódicas después de finalizar el tratamiento. A muchos pacientes que recaen no se les detecta la enfermedad mediante gammagrafías, sino por los síntomas que presentan. Factores como la estratificación del riesgo, los sitios de la enfermedad, los marcadores biomoleculares y la dosis de radiación acumulada tal vez se tengan en cuenta en la vigilancia tras el tratamiento.[
En una serie de 183 pacientes diagnosticados con neuroblastoma, 50 presentaron recidiva o progresión. Se detectó recaída de la enfermedad en la mayoría de los pacientes mediante síntomas o examen médico, gammagrafía con MIBG, catecolaminas urinarias y radiografías o ecografía.[
- De los 50 pacientes, 37 tenían enfermedad evidente desde el punto de vista clínico o cuantificable detectada mediante radiografía, ecografía o catecolaminas urinarias. Se identificaron 8 recidivas adicionales mediante la adición de gammagrafía MIBG.
- El diagnóstico por imágenes transversales (tomografía computarizada [TC]/resonancia magnética) solo fue necesario para identificar el 10 % de los casos (5 de 50).
- De las 50 recaídas, 32 se detectaron mediante investigaciones de vigilancia programadas, y 18 de las 50 (36 %) recaídas se detectaron debido a nuevos síntomas o a antecedentes médicos.
- De las 50 recaídas, 23 se relacionaron con nuevos síntomas preocupantes o con exámenes médicos. Como resultado, a 18 de los 50 pacientes se les realizó un diagnóstico por imagen antes de lo previsto, y 17 de ellos presentaban lesiones nuevas que se correspondían con los síntomas o el examen médico. De los 18 pacientes, 17 eran de riesgo alto en el momento del diagnóstico.
El uso de imágenes transversales con tomografía computarizada es controversial por la cantidad de radiación que emite y la baja proporción de recaídas que se detectan con esta modalidad.[
Referencias:
- Liang WH, Federico SM, London WB, et al.: Tailoring Therapy for Children With Neuroblastoma on the Basis of Risk Group Classification: Past, Present, and Future. JCO Clin Cancer Inform 4: 895-905, 2020.
- Cotterill SJ, Pearson AD, Pritchard J, et al.: Clinical prognostic factors in 1277 patients with neuroblastoma: results of The European Neuroblastoma Study Group 'Survey' 1982-1992. Eur J Cancer 36 (7): 901-8, 2000.
- Moroz V, Machin D, Faldum A, et al.: Changes over three decades in outcome and the prognostic influence of age-at-diagnosis in young patients with neuroblastoma: a report from the International Neuroblastoma Risk Group Project. Eur J Cancer 47 (4): 561-71, 2011.
- Look AT, Hayes FA, Shuster JJ, et al.: Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 9 (4): 581-91, 1991.
- Schmidt ML, Lukens JN, Seeger RC, et al.: Biologic factors determine prognosis in infants with stage IV neuroblastoma: A prospective Children's Cancer Group study. J Clin Oncol 18 (6): 1260-8, 2000.
- Berthold F, Trechow R, Utsch S, et al.: Prognostic factors in metastatic neuroblastoma. A multivariate analysis of 182 cases. Am J Pediatr Hematol Oncol 14 (3): 207-15, 1992.
- Matthay KK, Perez C, Seeger RC, et al.: Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children's Cancer Group study. J Clin Oncol 16 (4): 1256-64, 1998.
- Shimada H, Umehara S, Monobe Y, et al.: International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92 (9): 2451-61, 2001.
- Bagatell R, Beck-Popovic M, London WB, et al.: Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27 (3): 365-70, 2009.
- Canete A, Gerrard M, Rubie H, et al.: Poor survival for infants with MYCN-amplified metastatic neuroblastoma despite intensified treatment: the International Society of Paediatric Oncology European Neuroblastoma Experience. J Clin Oncol 27 (7): 1014-9, 2009.
- Attiyeh EF, London WB, Mossé YP, et al.: Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 353 (21): 2243-53, 2005.
- Spitz R, Hero B, Simon T, et al.: Loss in chromosome 11q identifies tumors with increased risk for metastatic relapses in localized and 4S neuroblastoma. Clin Cancer Res 12 (11 Pt 1): 3368-73, 2006.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Park JR, Kreissman SG, London WB, et al.: Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 322 (8): 746-755, 2019.
- Brodeur GM, Pritchard J, Berthold F, et al.: Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11 (8): 1466-77, 1993.
- Brodeur GM, Seeger RC, Barrett A, et al.: International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6 (12): 1874-81, 1988.
- Park JR, Bagatell R, Cohn SL, et al.: Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol 35 (22): 2580-2587, 2017.
- Bagatell R, McHugh K, Naranjo A, et al.: Assessment of Primary Site Response in Children With High-Risk Neuroblastoma: An International Multicenter Study. J Clin Oncol 34 (7): 740-6, 2016.
- Newman EA, Nuchtern JG: Recent biologic and genetic advances in neuroblastoma: Implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg 25 (5): 257-264, 2016.
- Overman RE, Kartal TT, Cunningham AJ, et al.: Optimization of percutaneous biopsy for diagnosis and pretreatment risk assessment of neuroblastoma. Pediatr Blood Cancer 67 (5): e28153, 2020.
- Gabra HO, Irtan S, Cross K, et al.: Minimally invasive surgery for neuroblastic tumours: A SIOPEN multicentre study: Proposal for guidelines. Eur J Surg Oncol 48 (1): 283-291, 2022.
- Zenitani M, Yoshida M, Matsumoto S, et al.: Feasibility and safety of laparoscopic tumor resection in children with abdominal neuroblastomas. Pediatr Surg Int 39 (1): 91, 2023.
- Chang S, Lin Y, Yang S, et al.: Safety and feasibility of laparoscopic resection of abdominal neuroblastoma without image-defined risk factors: a single-center experience. World J Surg Oncol 21 (1): 113, 2023.
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.
- Adkins ES, Sawin R, Gerbing RB, et al.: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39 (6): 931-6, 2004.
- Castel V, Tovar JA, Costa E, et al.: The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37 (11): 1574-8, 2002.
- La Quaglia MP, Kushner BH, Su W, et al.: The impact of gross total resection on local control and survival in high-risk neuroblastoma. J Pediatr Surg 39 (3): 412-7; discussion 412-7, 2004.
- Simon T, Häberle B, Hero B, et al.: Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31 (6): 752-8, 2013.
- Englum BR, Rialon KL, Speicher PJ, et al.: Value of surgical resection in children with high-risk neuroblastoma. Pediatr Blood Cancer 62 (9): 1529-35, 2015.
- von Allmen D, Davidoff AM, London WB, et al.: Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol 35 (2): 208-216, 2017.
- Mullassery D, Farrelly P, Losty PD: Does aggressive surgical resection improve survival in advanced stage 3 and 4 neuroblastoma? A systematic review and meta-analysis. Pediatr Hematol Oncol 31 (8): 703-16, 2014.
- Irtan S, Brisse HJ, Minard-Colin V, et al.: Image-defined risk factor assessment of neurogenic tumors after neoadjuvant chemotherapy is useful for predicting intra-operative risk factors and the completeness of resection. Pediatr Blood Cancer 62 (9): 1543-9, 2015.
- Wolden SL, Gollamudi SV, Kushner BH, et al.: Local control with multimodality therapy for stage 4 neuroblastoma. Int J Radiat Oncol Biol Phys 46 (4): 969-74, 2000.
- Hsu LL, Evans AE, D'Angio GJ: Hepatomegaly in neuroblastoma stage 4s: criteria for treatment of the vulnerable neonate. Med Pediatr Oncol 27 (6): 521-8, 1996.
- Katzenstein HM, Kent PM, London WB, et al.: Treatment and outcome of 83 children with intraspinal neuroblastoma: the Pediatric Oncology Group experience. J Clin Oncol 19 (4): 1047-55, 2001.
- Friedman DN, Goodman PJ, Leisenring WM, et al.: Long-Term Morbidity and Mortality Among Survivors of Neuroblastoma Diagnosed During Infancy: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (8): 1565-1576, 2023.
- De Bernardi B, Pianca C, Pistamiglio P, et al.: Neuroblastoma with symptomatic spinal cord compression at diagnosis: treatment and results with 76 cases. J Clin Oncol 19 (1): 183-90, 2001.
- Simon T, Niemann CA, Hero B, et al.: Short- and long-term outcome of patients with symptoms of spinal cord compression by neuroblastoma. Dev Med Child Neurol 54 (4): 347-52, 2012.
- McGirt MJ, Chaichana KL, Atiba A, et al.: Incidence of spinal deformity after resection of intramedullary spinal cord tumors in children who underwent laminectomy compared with laminoplasty. J Neurosurg Pediatr 1 (1): 57-62, 2008.
- Kraal K, Blom T, van Noesel M, et al.: Treatment and outcome of neuroblastoma with intraspinal extension: A systematic review. Pediatr Blood Cancer 64 (8): , 2017.
- Angelini P, Plantaz D, De Bernardi B, et al.: Late sequelae of symptomatic epidural compression in children with localized neuroblastoma. Pediatr Blood Cancer 57 (3): 473-80, 2011.
- De Bernardi B, Quaglietta L, Haupt R, et al.: Neuroblastoma with symptomatic epidural compression in the infant: the AIEOP experience. Pediatr Blood Cancer 61 (8): 1369-75, 2014.
- Papathanasiou ND, Gaze MN, Sullivan K, et al.: 18F-FDG PET/CT and 123I-metaiodobenzylguanidine imaging in high-risk neuroblastoma: diagnostic comparison and survival analysis. J Nucl Med 52 (4): 519-25, 2011.
- Kushner BH, Kramer K, Modak S, et al.: Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. J Clin Oncol 27 (7): 1041-6, 2009.
- Owens C, Li BK, Thomas KE, et al.: Surveillance imaging and radiation exposure in the detection of relapsed neuroblastoma. Pediatr Blood Cancer 63 (10): 1786-93, 2016.
Tratamiento del neuroblastoma de riesgo no alto
Neuroblastoma de riesgo bajo
El neuroblastoma de riesgo bajo representa casi la mitad de todos los pacientes con diagnóstico reciente. El éxito de los ensayos clínicos anteriores del Children's Oncology Group (COG) contribuyó a la reducción continua del tratamiento en determinados pacientes con neuroblastoma. De acuerdo con la categorización de riesgo del COG, los pacientes con enfermedad de riesgo bajo suelen presentar enfermedad en estadio bajo (estadio 1, 2A o 2B del International Neuroblastoma Staging System [INSS] y estadio L1 del International Neuroblastoma Risk Group [INRGSS]) y los tumores no presentan amplificación de MYCN, son hiperdiploides y tienen tipo histológico favorable. Para obtener más información sobre las categorías de riesgo del COG, consultar el
Para obtener más información sobre la enfermedad en estadio bajo, consultar la sección
Opciones de tratamiento del neuroblastoma de riesgo bajo
Un cirujano con experiencia debe resecar el tumor de los pacientes con enfermedad localizada que parece resecable (ausencia de factores de riesgo definidos por imágenes [L1] o según la pericia del cirujano). Si se confirma que las características biológicas son favorables después de la cirugía, la enfermedad residual no se considera un factor de riesgo de recaída y no se indica quimioterapia. En varios estudios se observó que los pacientes con características biológicas favorables y enfermedad residual tienen desenlaces excelentes, con tasas de supervivencia sin complicaciones (SSC) superiores al 90 % y tasas de supervivencia general (SG) que oscilan entre el 99 % y el 100 %.[
Se han observado sin hacer una biopsia algunos pacientes en los que se sospecha la presencia de neuroblastoma. El COG está estudiando está estrategia en más detalle en el ensayo ANBL1232 (NCT02176967) (no se aceptan nuevas inscripciones).[
Las opciones de tratamiento del neuroblastoma de riesgo bajo son las siguientes:
-
Cirugía seguida de observaciónCirugía seguida de observación (niños >1 año con estadio 1 o 2 del INSS). -
Observación con biopsia o sin estaObservación con biopsia o sin esta .- Observación sin biopsia (para neuroblastoma perinatal con tumores suprarrenales pequeños). La experiencia del COG con la observación de un neuroblastoma aparente sin hacer una biopsia diagnóstica es limitada y está en investigación.
- Observación con biopsia (para lactantes de <12 meses con enfermedad en estadio 4S sin hepatomegalia y tumores sin amplificación de MYCN; lactantes de <12 meses con enfermedad localizada, características histológicas y genómicas favorables y tumores sin amplificación de MYCN sin anomalías cromosómicas segmentarias).
-
Quimioterapia con cirugía o sin estaQuimioterapia con cirugía o sin esta (para enfermedad sintomática o enfermedad progresiva irresecable después de la cirugía). - Radioterapia (solo como tratamiento de urgencia).
Cirugía seguida de observación
La cirugía sola puede ser el tratamiento de pacientes en la categoría de riesgo bajo. Para obtener más información, consultar el
Evidencia (cirugía seguida de observación):
- En los resultados del estudio COG-P9641 se observó que la cirugía sola, incluso sin resección completa, cura a casi todos los pacientes con neuroblastoma en estadio 1 y a la gran mayoría de pacientes con enfermedad en estadios 2A o 2B del INSS que son asintomáticos y tienen características biológicas favorables.[
22 ] - En un ensayo clínico no aleatorizado en Japón se observaron resultados similares.[
55 ]
Observación con biopsia o sin esta
La observación sin biopsia se ha utilizado en el tratamiento del neuroblastoma perinatal con tumores suprarrenales pequeños.
En un estudio del COG se determinó que se pueden observar de manera segura masas suprarrenales pequeñas en estadio 1 o estadio 2 del INSS, que se sospecha que son neuroblastomas, en lactantes menores de 6 meses mediante ecografía de detección o incidental, sin necesidad de obtener un diagnóstico histológico definitivo ni de realizar una intervención quirúrgica. Mediante esta técnica se evitan las posibles complicaciones de la cirugía en pacientes recién nacidos.[
Evidencia (observación sin biopsia):
- En el estudio COG-ANBL00P2 se notificó que una conducta expectante de observación es inocua en pacientes menores de 6 meses con tumores suprarrenales sólidos que miden menos de 3,1 cm (o tumores quísticos de menos de 5 cm) y enfermedad en estadio 1 del INSS.[
33 ]- En este estudio, 67 de 83 pacientes (81 %) presentaron remisión espontánea y evitaron la intervención quirúrgica.
- De los 87 pacientes aptos, se observaron 83 sin biopsia ni resección y solo 16 (19 %) finalmente se sometieron a cirugía.
- La tasa de SSC a 3 años para un caso de neuroblastoma fue del 97,7 % y la tasa de SG fue del 100 %.
Existe polémica en cuanto a la necesidad de intentar una resección, en el momento del diagnóstico o después, en los lactantes asintomáticos de 12 meses de vida o menos con enfermedad en estadios 2B y 3, sin amplificación de MYCN y con características biológicas favorables. En un ensayo clínico alemán, se observaron algunos de estos pacientes después de una biopsia o una resección parcial sin quimioterapia ni radioterapia; muchos pacientes no presentaron progresión localizada y nunca se sometieron a resección adicional.[
Quimioterapia con cirugía o sin esta
La quimioterapia con cirugía o sin esta se usa para tratar los siguientes tipos de enfermedad:
- Enfermedad sintomática. El régimen de quimioterapia se compone de carboplatino, ciclofosfamida, doxorrubicina y etopósido. La dosis acumulada de cada fármaco quimioterapéutico se mantiene baja para reducir al mínimo los efectos a largo plazo.[
22 ] - Enfermedad progresiva irresecable después de la cirugía.
Evidencia (para la eliminación de la quimioterapia):
- El estudio COG-P9641 fue uno de los primeros estudios del COG en los que se probó la estratificación del riesgo a partir de factores derivados del consenso. En este ensayo no aleatorizado de fase III, se sometió a 915 lactantes y niños con enfermedad en estadio 2A y 2B del INSS a una operación inicial con el fin de obtener tejido para estudios diagnósticos y biológicos, y lograr la máxima resección inocua del tumor primario. Se reservó la quimioterapia para pacientes con enfermedad sintomática o en riesgo de tenerla, con una resección de menos del 50 % del tumor en el momento del diagnóstico o con enfermedad progresiva irresecable después de la cirugía sola.[
22 ]- Estadio 1:
- Los pacientes con enfermedad en estadio 1 lograron una tasa de SSC a 5 años del 93 % y una tasa de SG a 5 años del 99 %.
- Estadios 2A y 2B:
- Los pacientes asintomáticos con enfermedad en estadios 2A y 2B (n = 306), que se sometieron a observación después de la operación inicial, tuvieron una tasa de SSC a 5 años del 87 % y una tasa de SG a 5 años del 96 %.
- La tasa de SSC fue significativamente mejor para los pacientes de neuroblastoma en estadio 2A que para los pacientes en estadio 2B (92 vs. 85 %; P = 0,0321), pero la SG no difirió de modo significativo (98 vs. 96 %; P = 0,2867).
- Se cumplió con el objetivo principal del estudio (lograr una tasa de SG a 3 años del 95 % para los pacientes asintomáticos con enfermedad en estadios 2A y 2B).
- Los pacientes con enfermedad en estadio 2B tuvieron tasas de SSC y SG más bajas si tenían características histológicas desfavorables (tasa de SSC, 72 %; tasa de SG, 86 %), tumores diploides (tasa de SSC, 75 %; tasa de SG, 84 %) o más de 18 meses de edad.
- Los desenlaces para los pacientes con tumores diploides en estadio 2B y características histológicas desfavorables fueron en especial precarios (tasa de SSC, 54 %; tasa de SG, 70 %), sin sobrevivientes entre los pocos pacientes que, además, tenían pérdida de heterocigosis en 1p.
- Todas las muertes fueron de niños mayores de 18 meses.
- Desenlace de los pacientes asintomáticos en el momento del diagnóstico que se sometieron a observación después de la operación inicial y de los pacientes que se trataron con quimioterapia tras la operación: de los 915 pacientes del grupo de la operación inicial, 800 eran asintomáticos en el momento del diagnóstico y se sometieron a observación después de la operación. Dentro de este grupo, el 11 % de los pacientes presentaron enfermedad recidivante o progresiva. De los 115 pacientes que se sometieron a cirugía seguida de quimioterapia inmediata (mediana, 4 ciclos; intervalo, 1 a 8), el 81 % de los pacientes presentaron una respuesta parcial muy buena o mejor. Después de la quimioterapia, el 10 % de los pacientes presentaron recidiva o progresión de la enfermedad.
- En los pacientes que se sometieron a cirugía sola, la tasa de SSC a 5 años fue del 89 %, y la SG estimada fue del 97 %.
- En los pacientes que se sometieron a cirugía y quimioterapia inmediata, la tasa de SSC a 5 años fue del 91 % y la SG estimada fue del 98 %.
- Amplificación de MYCN: se analizó el efecto de los tumores con amplificación de MYCN en pacientes con enfermedad en estadio 1.
- Para los pacientes con tumores sin amplificación de MYCN, la tasa de SSC a 5 años fue del 93 % y la tasa de SG fue del 99 %.
- Para los pacientes con tumores con amplificación de MYCN, la tasa de SSC a 5 años fue del 70 % (P = 0, 0042), y la tasa de SG fue del 80 % (P = 0, 001).
- Estadio 1:
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el
Ensayos clínicos en curso
Realizar una
Neuroblastoma de riesgo intermedio
Según la clasificación de riesgos del COG, en el riesgo intermedio se incluyen los siguientes pacientes:
- Pacientes con enfermedad en estadio 2A o 2B, con resección inferior al 50 % o biopsia solo de tumores sin amplificación de MYCN.
- Pacientes con enfermedad sin amplificación de MYCN en estadio 2A/2B o 4S sintomáticos.
- Niños menores de 18 meses con enfermedad en estadio 3 sin amplificación de MYCN (con independencia de las características histológicas).
- Niños mayores de 18 meses con enfermedad en estadio 3, sin amplificación de MYCN y características histológicas favorables.
- Lactantes menores de 12 meses con enfermedad en estadio 4, sin amplificación de MYCN y con características histológicas favorables.
- Lactantes de 12 a 18 meses con tumores en estadio 4, sin amplificación de MYCN, características histológicas favorables y tumores hiperdiploides.
Se incluyó un subconjunto de pacientes en estadio 4S y se clasificaron como de riesgo intermedio si tenían tumores diploides o características histológicas desfavorables y sin amplificación de MYCN.
Para obtener más información sobre las categorías de riesgo del COG, consultar el
Los resultados del estudio COG-A3961 (NCT00003093) de riesgo intermedio,[
Para obtener más información sobre el esquema de clasificación pretratamiento para el neuroblastoma de riesgo intermedio, consultar la sección
Para obtener más información sobre los tumores en estadios 4S y MS, consultar la sección
Opciones de tratamiento del neuroblastoma de riesgo intermedio
Las opciones de tratamiento del neuroblastoma de riesgo intermedio son las siguientes:
-
Quimioterapia con cirugía o sin estaQuimioterapia con cirugía o sin esta . -
Cirugía y observaciónCirugía y observación (para lactantes). -
RadioterapiaRadioterapia (si fuera necesaria).
Quimioterapia con cirugía o sin esta
Los pacientes de la categoría de riesgo intermedio se tratan con éxito mediante resección quirúrgica completa y 2, 4 u 8 ciclos de quimioterapia neoadyuvante. El régimen de quimioterapia se compone de carboplatino, ciclofosfamida, doxorrubicina y etopósido, y la dosis acumulada de cada fármaco se mantiene baja para reducir al mínimo los efectos a largo plazo del régimen de quimioterapia (ANBL0531 [NCT00499616]). Por regla general, los pacientes cuyos tumores tenían características biológicas desfavorable recibieron 8 ciclos de quimioterapia, en lugar de los 2 o 4 ciclos administrados a los pacientes cuyos tumores tenían características biológicas favorables.
En los casos de neuroblastoma abdominal en los que se piensa que hay compromiso renal, no se realiza una nefrectomía antes de la administración de un curso de quimioterapia.[
Son necesarios más estudios para determinar si la quimioterapia inicial está indicada para todos los lactantes con neuroblastoma localizado de riesgo intermedio.
Evidencia (quimioterapia con cirugía o sin esta):
- El objetivo del estudio ANBL0531 (NCT00499616) fue reducir el tratamiento de subgrupos de pacientes con neuroblastoma de riesgo intermedio (sin amplificación de MYCN, edad y estadio definidos). La duración del tratamiento (2, 4 u 8 ciclos de quimioterapia neoadyuvante a dosis moderadas) se asignó en función de las características clínicas y de un algoritmo basado en la respuesta y la biología del tumor (incluido el estado alélico de 1p36 y 11q23). Las tasas de SSC y SG a 3 años de toda la cohorte del estudio (N = 404) fueron del 83,2 % y el 94,9 %, respectivamente. La duración y la intensidad del tratamiento se redujeron en varios subconjuntos de pacientes. En el estudio se añadieron pacientes en estadio 4 con características biológicas favorables que tenían entre 12 y 18 meses de edad.[
77 ]- En el estudio preexistente (A3961 [NCT00003093]), la administración de la quimioterapia neoadyuvante facilitó, al menos, una resección parcial en el 99,6 % de los tumores que antes se consideraban irresecables. No se observaron diferencias significativas en la SG de acuerdo con el grado de resección obtenido (completa vs. incompleta).[
66 ,77 ] - Menos del 3 % de los pacientes del estudio ANBL0531 recibieron radioterapia local, y solo los pacientes con agrandamiento hepático progresivo o compresión de la médula espinal recibieron radioterapia.[
77 ] - La tasa de SSC a 3 años fue del 92 % para los pacientes con enfermedad en estadio 3 y tipo histopatológico favorable (n = 269); del 90 % para los pacientes con enfermedad en estadio 4S y características biológicas desfavorables, incluso diploidía o tipo histológico desfavorable (n = 31), y del 81 % para los lactantes con enfermedad en estadio 4 (n = 176) (P < 0,001 para los estadios 3 y 4S vs. el estadio 4).
- Los lactantes con enfermedad en estadio 4 con características biológicas favorables (n = 61) tuvieron una SSC a 3 años superior, en comparación con aquellos que presentaron tumores confirmados con características biológicas desfavorables (n = 47) (tasa de SSC a 3 años, 86,9 vs. 66,8 %; P = 0,02), aunque la SG no fue significativamente diferente (tasa de SG a 3 años, 95,0 vs. 86,7 %; P = 0,08).
- Solo se estratificó a los lactantes según la ploidía; aquellos con tumores diploides recibieron 8 ciclos de quimioterapia en lugar de 4. Las tasas estimadas de SG a 3 años fueron del 98 % para la enfermedad en estadio 3, del 97 % para la enfermedad en estadio 4S y del 93 % para la enfermedad en estadio 4 (P = 0,002 para los estadios 3 y 4S vs. el estadio 4). Los lactantes con diploidía tuvieron un desenlace más precario (P = 0,03), al igual que el grupo combinado de todos los pacientes con diploidía (P = 0,03).
- No hubo ninguna diferencia en la SG en pacientes con características biológicas favorables entre los que recibieron 8 ciclos de quimioterapia (100 %) para una enfermedad persistente, en comparación con aquellos que recibieron 4 ciclos (96 %).
- No se presentaron efectos tóxicos inesperados.
- En el estudio preexistente (A3961 [NCT00003093]), la administración de la quimioterapia neoadyuvante facilitó, al menos, una resección parcial en el 99,6 % de los tumores que antes se consideraban irresecables. No se observaron diferencias significativas en la SG de acuerdo con el grado de resección obtenido (completa vs. incompleta).[
- En un ensayo clínico prospectivo alemán participaron 340 lactantes de 1 año o menos con tumores en estadios 1, 2 o 3, verificados histológicamente y sin amplificación de MYCN. En el momento del diagnóstico, se administró quimioterapia a 57 lactantes con órganos amenazados por el tumor. El tumor se resecó en forma total o subtotal en 190 lactantes que se sometieron a cirugía de riesgo bajo. Se observó, sin administrar quimioterapia, a 93 lactantes cuyos tumores solo se podían resecar con cirugía de riesgo alto, debido a la edad o el compromiso orgánico.[
44 ]- La tasa de SG a 3 años fue excelente (95 %) para los lactantes que recibieron quimioterapia.
- Se evitó realizar más cirugías a 33 lactantes y se evitó la quimioterapia en 72 lactantes.
- La tasa de SG a 3 años para los lactantes sometidos a observación sin tratamiento fue del 99 %. La tasa de supervivencia sin metástasis fue del 94 % para los lactantes con tumores no resecados y no fue diferente a la de los lactantes tratados con cirugía o quimioterapia (mediana de seguimiento, 58 meses).
- De los 93 lactantes con tumores no resecados, 44 presentaron una remisión espontánea (17 fueron remisiones completas) y 39 lactantes presentaron progresión.
- Los investigadores indicaron que una conducta expectante es adecuada para lactantes con neuroblastoma localizado porque se han observado remisiones después del primer año de vida.
- La quimioterapia de dosis moderada demostró ser eficaz en el ensayo prospectivo Infant Neuroblastoma European Study (EURO-INF-NB-STUDY-1999-99.1). Cerca de la mitad de los lactantes con neuroblastoma irresecable no metastásico y sin amplificación de MYCN se sometieron a una resección quirúrgica inocua y evitaron efectos adversos a largo plazo.[
99 ][Nivel de evidencia C1]- La tasa de SG a 5 años fue del 99 % y la tasa de SSC fue del 90 % (mediana de seguimiento, 6 años).
- En este estudio, los lactantes sometidos a resección quirúrgica tuvieron mejor SSC que aquellos que no se sometieron a cirugía.
- En un ensayo prospectivo de la International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN), se trató a lactantes con neuroblastoma irresecable en estadio 2 o 3, sin amplificación de MYCN y niños de 12 a 18 meses de edad con características favorables según la International Neuroblastoma Pathology Classification.[
1010 ][Nivel de evidencia C2]- La tasa de SSC fue del 98 % con quimioterapia convencional.
- Estos resultados son similares a los resultados del ensayo COG-A3961.
- En dos ensayos prospectivos europeos con lactantes que tenían neuroblastoma diseminado sin amplificación del gen MYCN, no se administró quimioterapia a los lactantes con enfermedad en estadio 3 del INSS o centellografía esquelética positiva sin metástasis ósea radiológica (identificada principalmente por gammagrafía con MIBG, unos pocos solo por gammagrafía ósea con tecnecio Tc99m), a menos que presentaran síntomas que pusieran en peligro su vida u órganos. Cuando se administró quimioterapia, se usaron dosis bajas y estándar.[
1111 ]- La tasa de SG fue del 100 % en los 41 pacientes que no tenían enfermedad en estadio 4S del INSS, con independencia de la quimioterapia inicial.
- En los lactantes con metástasis evidentes en el esqueleto, los pulmones y el sistema nervioso central (por gammagrafía con radionúclidos, pero no por una radiografía común o tomografía computarizada [TC]), la tasa de SG a 2 años fue del 96 % (n = 45).
- Ningún paciente murió por complicaciones relacionadas con la cirugía o la quimioterapia en ninguno de los protocolos.
- En un análisis retrospectivo del COG se evaluó a pacientes de 12 y 18 meses de edad con enfermedad metastásica y características biológicas favorables. En los ensayos preexistentes, estos pacientes recibieron tratamiento de la enfermedad con regímenes de riesgo alto.[
1212 ]- En este análisis se demostró que este grupo de pacientes tuvo resultados similares excelentes con la terapia de riesgo intermedio, en comparación con los de la terapia de riesgo alto. En la actualidad, estos pacientes se tratan con terapia de riesgo intermedio en los ensayos clínicos en curso.
Cirugía y observación (para lactantes)
La necesidad de quimioterapia para todos los lactantes asintomáticos con enfermedad en estadios 3 o 4 resulta algo polémica porque en algunos estudios europeos se observaron resultados favorables con cirugía y observación.[
Evidencia (cirugía y observación en lactantes):
- En un estudio francés se notificó que los lactantes clasificados en estadio 4, debido a un tumor primario infiltrante más allá de la línea media (primario en estadio 3 del INSS con metástasis limitadas a la categoría 4S) o con centellografía ósea positiva no relacionada con cambios de hueso cortical documentados mediante radiografías simples o TC.[
1313 ]- Tienen un mejor desenlace con quimioterapia menos intensiva que otros lactantes en estadio 4 (tasa de SSC, 90 vs. 27 %).
- Sin embargo, una proporción mucho más alta de aquellos con lesiones óseas corticales, según se demostró radiológicamente, también tenían tumores con amplificación de MYCN.
- A partir del estudio francés, la SIOPEN llevó a cabo un ensayo prospectivo con 125 lactantes (n = 41 con tumores primarios en estadio 3 del INSS o centellografía positiva) con neuroblastoma diseminado sin amplificación de MYCN para verificar si estos pacientes se podían someter a observación ante la ausencia de síntomas. Sin embargo, los médicos no siempre usaron una conducta expectante.[
1111 ]- No hubo ninguna diferencia significativa en las tasas de SG a 2 años entre los pacientes con tumores primarios irresecables y los pacientes con tumores primarios resecables (97 vs. 100 %), ni entre los pacientes con centellografía ósea negativa y positiva, sin anomalías radiológicas (100 vs. 97 %).
- En un ensayo clínico prospectivo alemán participaron 340 lactantes de 1 año o menos con tumores en estadios 1, 2 o 3, verificados histológicamente y sin amplificación de MYCN. De los 190 lactantes sometidos a resección, 8 tenían enfermedad en estadio 3. Se observó, sin administrar quimioterapia, a 93 lactantes cuyos tumores solo se podía resecar con una cirugía de riesgo alto debido a la edad o compromiso orgánico; 21 de ellos tenían enfermedad en estadio 3. De los lactantes, 57, incluso 41 pacientes en estadio 3, se trataron con quimioterapia para controlar síntomas amenazantes.[
44 ]- La tasa de SG a 3 años fue excelente en todo el grupo de lactantes con tumores no resecados (99 %), en los lactantes sometidos a quimioterapia (95 %) y en los lactantes con tumores resecados (98 %) (P = 0,45).
Radioterapia
La radioterapia para los niños con enfermedad de riesgo intermedio se reserva para los pacientes que presentan enfermedad progresiva durante el tratamiento con quimioterapia, o enfermedad progresiva irresecable después del tratamiento con quimioterapia.
En un ensayo aleatorizado prospectivo del COG, en el que se evaluó la quimioterapia de intensidad reducida para los pacientes con neuroblastoma de riesgo intermedio, solo 12 de 479 pacientes (2,5 %) recibió radioterapia local (21 Gy). Un paciente tenía enfermedad en estadio 4S; 5 pacientes, enfermedad en estadio 3; y 6 pacientes, enfermedad en estadio 4. La radioterapia se administró a los pacientes con deterioro clínico, sin importar el tipo de tratamiento inicial (8 pacientes), a los pacientes con enfermedad residual macroscópica y características biológicas desfavorables (3 pacientes) y a los pacientes con recaída después de la terapia (1 paciente).[
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el
Ensayos clínicos en curso
Realizar una
Referencias:
- Matthay KK, Perez C, Seeger RC, et al.: Successful treatment of stage III neuroblastoma based on prospective biologic staging: a Children's Cancer Group study. J Clin Oncol 16 (4): 1256-64, 1998.
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.
- Nuchtern JG, London WB, Barnewolt CE, et al.: A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg 256 (4): 573-80, 2012.
- Hero B, Simon T, Spitz R, et al.: Localized infant neuroblastomas often show spontaneous regression: results of the prospective trials NB95-S and NB97. J Clin Oncol 26 (9): 1504-10, 2008.
- Iehara T, Hamazaki M, Tajiri T, et al.: Successful treatment of infants with localized neuroblastoma based on their MYCN status. Int J Clin Oncol 18 (3): 389-95, 2013.
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- Shamberger RC, Smith EI, Joshi VV, et al.: The risk of nephrectomy during local control in abdominal neuroblastoma. J Pediatr Surg 33 (2): 161-4, 1998.
- Rubie H, De Bernardi B, Gerrard M, et al.: Excellent outcome with reduced treatment in infants with nonmetastatic and unresectable neuroblastoma without MYCN amplification: results of the prospective INES 99.1. J Clin Oncol 29 (4): 449-55, 2011.
- Kohler JA, Rubie H, Castel V, et al.: Treatment of children over the age of one year with unresectable localised neuroblastoma without MYCN amplification: results of the SIOPEN study. Eur J Cancer 49 (17): 3671-9, 2013.
- De Bernardi B, Gerrard M, Boni L, et al.: Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol 27 (7): 1034-40, 2009.
- Bender HG, Irwin MS, Hogarty MD, et al.: Survival of Patients With Neuroblastoma After Assignment to Reduced Therapy Because of the 12- to 18-Month Change in Age Cutoff in Children's Oncology Group Risk Stratification. J Clin Oncol 41 (17): 3149-3159, 2023.
- Minard V, Hartmann O, Peyroulet MC, et al.: Adverse outcome of infants with metastatic neuroblastoma, MYCN amplification and/or bone lesions: results of the French society of pediatric oncology. Br J Cancer 83 (8): 973-9, 2000.
- Kim C, Choi YB, Lee JW, et al.: Excellent treatment outcomes in children younger than 18 months with stage 4 MYCN nonamplified neuroblastoma. Korean J Pediatr 61 (2): 53-58, 2018.
Tratamiento del neuroblastoma de riesgo alto
Los pacientes con mayor riesgo de progresión de la enfermedad y mortalidad tienen más de 18 meses de edad, presentan enfermedad metastásica o localizada con características biológicas desfavorables, como amplificación de MYCN, o tienen características histológicas desfavorables. Para obtener más información sobre las categorías de riesgo del Children's Oncology Group (COG), consultar el
Alrededor del 8 al 10 % de los lactantes con enfermedad en estadio MS tienen tumores con amplificación de MYCN y se suelen tratan mediante protocolos de riesgo alto. Las tasas de supervivencia sin complicaciones (SSC) y supervivencia general (SG) a 5 años fueron del 60 % y el 64 %, respectivamente, para los lactantes con enfermedad en estadio MS y amplificación de MYCN (n = 23), entre los 5000 pacientes inscritos en el ensayo del COG ANBL00B1 (NCT00904241).[
Para los niños con neuroblastoma de riesgo alto que recibieron los tratamientos actuales, la tasa de SG a 5 años fue de alrededor del 60 % para los pacientes diagnosticados entre 2007 y 2017.[
En un estudio de la base de datos del International Neuroblastoma Risk Group (INRG), se identificó a 146 pacientes con metástasis a distancia limitada a los ganglios linfáticos (denominadas en estadio 4N), con tendencia a presentar enfermedad con características biológicas favorables y desenlace favorable (tasa de SG a 5 años, 85 %). Este hallazgo indica que, para este subgrupo especial y muy infrecuente de pacientes de riesgo alto en estadio 4, podría considerarse la administración de un tratamiento menos intensivo.[
Opciones de tratamiento del neuroblastoma de riesgo alto
Los desenlaces para pacientes con neuroblastoma de riesgo alto siguen siendo precarios, a pesar de las mejoras recientes en la supervivencia observadas en ensayos aleatorizados.
Las opciones de tratamiento del neuroblastoma de riesgo alto suelen ser las siguientes:
-
Régimen de quimioterapia, cirugía, ciclos en tándem de terapia mielosupresora y trasplante de células madre hematopoyéticas (TCMH), radioterapia y dinutuximab con factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) e isotretinoínaRégimen de quimioterapia, cirugía, ciclos en tándem de terapia mielosupresora y trasplante de células madre hematopoyéticas (TCMH), radioterapia y dinutuximab con factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) e isotretinoína .
Quimioterapia, cirugía, ciclos en tándem de terapia mielosupresora y TCMH, radioterapia y dinutuximab con GM-CSF e isotretinoína.
Por lo general, el tratamiento de pacientes con enfermedad de riesgo alto se divide en las siguientes 3 fases:
-
InducciónInducción (incluye quimioterapia y resección quirúrgica). -
ConsolidaciónConsolidación (ciclos en tándem de terapia mielosupresora y TCMH, y radioterapia dirigida al sitio del tumor primario y a los sitios metastásicos residuales). -
PosconsolidaciónPosconsolidación (inmunoterapia con GM-CSF y tratamiento con isotretinoína).
Fase de inducción
El tratamiento de base que más se usa como terapia de inducción incluye ciclos de dosis intensivas de cisplatino y etopósido, alternados con vincristina, ciclofosfamida y doxorrubicina.[
Evidencia (quimioterapia de inducción con tratamientos adicionales o sin estos):
- En un estudio, la adición del tratamiento con dinutuximab anti-GD2 con GM-CSF y dosis bajas de interleucina-2 (IL-2) administradas con cada curso de quimioterapia de inducción, produjo desenlaces alentadores en 42 niños con enfermedad en estadio 4 recién diagnosticada.[
1111 ]- Esta terapia de inducción, seguida de una terapia estándar de consolidación y posconsolidación produjo respuestas parciales tempranas o mejores en la mayoría de los pacientes, redujo los volúmenes tumorales y obtuvo una alentadora tasa de SSC a 3 años del 73,7 %.
- En un ensayo europeo prospectivo controlado y aleatorizado se investigó la terapia de inducción prolongada en 422 pacientes con neuroblastoma de riesgo alto recién diagnosticado. Los pacientes se asignaron de manera aleatoria para recibir quimioterapia de inducción estándar con 6 cursos de quimioterapia o quimioterapia de inducción experimental que comenzó con 2 cursos adicionales de topotecán, ciclofosfamida y etopósido seguidos de quimioterapia de inducción estándar.[
1212 ][Nivel de evidencia B3]- La tasa de SSC a 3 años fue del 34 % en los pacientes que recibieron el régimen de inducción experimental y del 32 % en los pacientes que recibieron el régimen de inducción estándar.
- La adición de 2 cursos de quimioterapia con topotecán no mejoró la SSC de los pacientes con neuroblastoma de riesgo alto y produjo más efectos tóxicos por paciente.
- Investigadores europeos completaron otro estudio aleatorizado de regímenes de inducción para pacientes con neuroblastoma de riesgo alto. Un total de 630 pacientes se asignaron de manera aleatoria para recibir cisplatino, vincristina, carboplatino, etopósido y ciclofosfamida (régimen rCOJEC; n = 313) o los regímenes de inducción N5 del Memorial Sloan Kettering Cancer Center (MSKCC-N5; n = 317).[
1313 ][Nivel de evidencia B1]- No hubo diferencias significativas en las tasas de respuesta metastásica completa entre los dos regímenes (32 % para rCOJEC vs. 35 % para MSKCC-N5; P = 0,368) ni en las tasas de SSC a 3 años (44 % para rCOJEC vs. 47 % para MSKCC-N5; P = 0,527).
- Los pacientes que recibieron el régimen rCOJEC presentaron menos efectos tóxicos agudos.
- Se ha seleccionado el régimen rCOJEC como el régimen de inducción estándar para el próximo ensayo de la International Society of Pediatric Oncology European Neuroblastoma (SIOPEN).
Después de una respuesta a la quimioterapia de inducción, por lo general, se intenta resecar el tumor primario. Es debatible si la resección macroscópica total es beneficiosa antes o después de la quimioterapia de inducción.[
Evidencia (resección del tumor primario antes o después de la quimioterapia):
- En el estudio del COG A3973 (NCT00004188) se hizo una revisión quirúrgica centralizada de 220 pacientes en quienes se intentó la resección macroscópica total después de la quimioterapia de inducción. Según el cálculo del cirujano, el grado de la resección se determinó como de un 90 % o más versus menos del 90 %, pero la concordancia solo fue del 63 % durante la revisión central con imágenes.[
1515 ][Nivel de evidencia C1]- Sin embargo, la evaluación del cirujano de una resección del 90 % o más versus menos del 90 % predijo una tasa de SSC del 46 % versus el 38 % (P = 0,01), respectivamente, y una incidencia acumulada de la tasa de recidiva local del 8,5 % versus el 20 %, respectivamente.
- No hubo ninguna diferencia significativa en las tasas de SG entre los 2 grupos (57 vs. 49 %, P = 0,3).
- La conclusión del autor sustenta la continuidad de la iniciativa de lograr una resección mayor del 90 % para disminuir la recidiva local.
- En un estudio retrospectivo de un solo centro con 87 niños de neuroblastoma de riesgo alto, no se observó ningún beneficio significativo de la resección macroscópica total, en comparación con la resección subtotal (>90 %).[
1616 ][Nivel de evidencia C2]- No obstante, los resultados indican que la resección de más del 90 % se relaciona con una mejora de la SG, en comparación con una resección de menos del 90 %.
No se ha demostrado, de manera inequívoca, el posible beneficio de los abordajes quirúrgicos radicales para pacientes de riesgo alto con enfermedad metastásica con el fin de lograr la resección completa del tumor, en el momento del diagnóstico o después de la quimioterapia. En varios estudios se notificó que la resección completa del tumor primario en el momento del diagnóstico mejoró la supervivencia; sin embargo, el desenlace en estos pacientes depende más de las características biológicas del tumor, que a veces determinan la resecabilidad, y no tanto del alcance de la resección quirúrgica.[
Respecto a los pacientes en estadio 4 mayores de 18 meses, hay polémica sobre si hay alguna ventaja de la resección macroscópica total del tumor primario después de la quimioterapia.[
Al final del tratamiento de inducción, los pacientes con enfermedad de riesgo alto suelen someterse a una evaluación completa de la enfermedad. El tratamiento de los pacientes con enfermedad residual al final de la terapia de inducción convencional no está estandarizado. En un estudio retrospectivo se analizó a 201 pacientes con enfermedad de riesgo alto que presentaron una respuesta parcial o inferior al final de la terapia de inducción. Los pacientes fueron seleccionados para recibir de inmediato quimioterapia de dosis alta (cohorte 1), terapia puente (a menudo quimioinmunoterapia de 131 I-MIBG) seguida de quimioterapia de dosis alta (cohorte 2), o terapia adicional, pero no quimioterapia de dosis alta (cohorte 3).[
- A pesar de tener características menos favorables, los pacientes de la cohorte 2 tuvieron una SSC similar a la de los pacientes de la cohorte 1, mientras que los pacientes de la cohorte 3 tuvieron una SSC inferior.
- Entre los pacientes con enfermedad estable en sitios metastásicos al final del tratamiento de inducción, los pacientes de la cohorte 2 tuvieron una SSC superior a la de los pacientes de la cohorte 1.
Estos datos retrospectivos indican una función de la terapia puente en pacientes con respuesta incompleta a la terapia de inducción convencional.
Fase de consolidación
Para la fase de consolidación de los regímenes de riesgo alto se usa la quimioterapia mielosupresora y el TCMH, en un intento por erradicar la enfermedad residual mínima (ERM) con dosis de quimioterapia supresora, que de otra manera serían mortales, luego se hace un rescate con células madre autógenas (recogidas durante la quimioterapia de inducción) con el fin de regenerar la médula ósea. En varios estudios aleatorizados controlados grandes se observó una mejora en las tasas de SSC a 3 años con el tratamiento de TCMH (31–47 %) versus la quimioterapia convencional (22–31 %).[
Evidencia (quimioterapia mielosupresora y rescate de células madre):
- En un ensayo multicéntrico europeo grande de terapia de consolidación se asignó al azar a los pacientes que habían finalizado un régimen de inducción con varios fármacos (cisplatino, carboplatino, ciclofosfamida, vincristina y etopósido con topotecán, vincristina y doxorrubicina o sin estos) y que lograron una respuesta adecuada a la administración de busulfano y melfalán o carboplatino, etopósido y melfalán.[
2828 ][Nivel de evidencia A1]- El tratamiento de inducción con cisplatino, carboplatino, ciclofosfamida, vincristina y etopósido, y la consolidación para el TCMH con busulfano y melfalán mejoró la SSC pero no afectó la SG y no produjo efectos secundarios graves.
- En un estudio clínico aleatorizado (COG-ANBL0532) se evaluó la eficacia de 2 ciclos versus 1 ciclo, de quimioterapia mielosupresora con rescate de células madre.[
2929 ][Nivel de evidencia A1] Los niños mayores de 18 meses con neuroblastoma en estadio 4, que recibieron antes 6 ciclos de quimioterapia de inducción, se asignaron al azar a recibir un solo TCMH autógeno con carboplatino, etopósido y melfalán o trasplantes en tándem con ciclofosfamida y tiotepa, seguido de una dosis reducida de carboplatino, etopósido y melfalán. Después de la radioterapia dirigida al lecho tumoral, la mayoría de los pacientes se asignaron al azar a otro ensayo separado para recibir isotretinoína sola o isotretinoína con dinutuximab y refuerzo inmunitario.- La tasa de SSC a 3 años desde el momento de la aleatorización fue del 62 % para los trasplantes en tándem y del 48 % para un solo TCMH (P = 0,006). La tasa de SG a 3 años fue del 74 % para los TCMH autógenos en tándem y del 69 % para un solo TCMH autógeno (P = 0,25).
- La tasa de SSC a 3 años en los pacientes aleatorizados que después recibieron dinutuximab y refuerzo inmunitario fue del 73 % para los TCMH en tándem y del 55 % para un solo TCMH (P = 0,004), mientras que la tasa de SG fue del 84 % y del 74 %, respectivamente.[
2929 ][Nivel de evidencia B1] - Los resultados de estos estudios tienen una limitación importante: no se hizo ninguna asignación aleatoria al tratamiento para un número considerable de pacientes inscritos en el estudio (debido a la preferencia del paciente o del proveedor de atención de la salud), lo que introduce un posible sesgo de selección.
- En una revisión de Cochrane actualizada se evaluaron 3 ensayos clínicos aleatorizados en los que se compararon el trasplante autógeno de médula ósea (TAMO) con la quimioterapia estándar.[
2424 ,2525 ,2626 ,3030 ,3131 ]- La SSC fue significativamente mejor con el trasplante autógeno de médula ósea (TAMO), pero no hubo diferencias estadísticamente significativas en la SG.
- En una revisión de 147 casos de trasplante alogénico enviados al Center for International Blood and Marrow Transplant Research no se observó ningún beneficio del trasplante alogénico sobre el autógeno, incluso si la persona receptora del trasplante alogénico había recibido un trasplante autógeno previo.[
3232 ]
En otro estudio prospectivo y aleatorizado, no se observó ningún beneficio de la purificación de las células madre obtenidas de las células de neuroblastoma antes del trasplante.[
Para obtener más información sobre trasplantes, consultar
Después de la terapia mielosupresora, se indica radiación dirigida al tumor primario (con independencia de si se realizó una escisión completa o no).[
Evidencia (radioterapia con refuerzo vs. radioterapia sin refuerzo para la resección incompleta):
- Debido a las tasas altas de recidiva local después de la resección quirúrgica incompleta, en el ensayo del COG ANBL0532 (NCT00567567) se evaluó de manera prospectiva el potencial de beneficio de la radioterapia de refuerzo para los pacientes con tumor residual macroscópico y se compararon los resultados con los del anterior ensayo clínico del COG sobre el neuroblastoma de riesgo alto (A3973 [NCT00004188]), en el que los pacientes no recibieron radioterapia de refuerzo. Todos los pacientes en el ensayo ANBL0532 recibieron 21,6 Gy de radiación dirigida al volumen preoperatorio del tumor primario después de la quimioterapia de inducción.[
3737 ][Nivel de evidencia C1]- No hubo diferencias en los desenlaces de los pacientes tratados en el ensayo ANBL0532 que recibieron un solo TCMH y radioterapia de refuerzo (n = 74) y los pacientes tratados en el ensayo A3973 que se sometieron a una resección incompleta y no recibieron radioterapia de refuerzo (n = 47).
- La incidencia acumulada a 5 años de progresión local fue del 16,3 % para los pacientes en el ensayo ANBL0532 versus el 10,6 % para los pacientes en el ensayo A3973 (P = 0,4126).
- La tasa de SSC fue del 50,9 % para los pacientes en el ensayo ANBL0532 versus el 48,9 % para los pacientes en el ensayo A3973 (P = 0,5084).
- La tasa de SG fue del 68,1 % para los pacientes en el ensayo ANBL0532 versus el 56,9 % para los pacientes en el ensayo A3973 (P = 0,2835).
- La radioterapia de refuerzo administrada en el tumor residual macroscópico que estaba presente al final de la inducción no mejoró de manera significativa la incidencia acumulada de progresión local a 5 años; por lo tanto, no se recomienda.[
3737 ]
La irradiación extensa de los ganglios linfáticos, con independencia del alcance de la resección quirúrgica previa al TCMH, no benefició a los pacientes en cuanto a la progresión local o la SG.[
El tratamiento de la enfermedad metastásica ósea, administrado en el momento de la irradiación del lecho tumoral primario, también se tiene en cuenta para maximizar el control de la enfermedad. La administración de radioterapia dirigida a sitios metastásicos se determina caso por caso, o según las directrices del protocolo para los pacientes inscritos en los estudios. Muchos niños presentan metástasis óseas generalizadas. Debido a que no es factible irradiar todos los sitios iniciales, la práctica actual es tratar los sitios que no han respondido, según los resultados de la MIBG antes del TCMH.[
En una serie retrospectiva de 159 niños con neuroblastoma en estadio M de riesgo alto, se administró irradiación focal a todos los sitios metastásicos, con independencia de la respuesta a la quimioterapia, a menos que las metástasis fueran demasiado numerosas.[
- La tasa de control a 5 años de los sitios metastásicos irradiados fue del 81 %.
- Las metástasis que se volvieron negativas para MIBG después de la quimioterapia tenían una probabilidad significativamente menor de recidiva que los sitios que siguieron siendo positivos para MIBG.
- Los pacientes cuya enfermedad no recidivó en las localizaciones metastásicas irradiadas tuvieron una SG mejor.
- Cuando fue factible administrar radioterapia, incluso en sitios que se resolvieron con quimioterapia de inducción, la radioterapia tuvo una eficacia superior al 90 % a la hora de proporcionar control de la enfermedad en esos sitios metastásicos.
Estas observaciones sustentan el paradigma actual de irradiación de las metástasis que siguen captando MIBG tras la quimioterapia de inducción en los pacientes de riesgo alto. No se aconseja irradiar más del 50 % de la médula ósea.[
En casos en los que hay metástasis óseas difusas persistentes después de la quimioterapia de inducción, se hace una reevaluación al terminar las dosis altas de quimioterapia antes de decidir la radioterapia de consolidación.
Se publicaron resultados preliminares del tratamiento de radioterapia con haz de protones para tratar pacientes con tumores primarios de neuroblastoma de riesgo alto, que reflejaron niveles aceptables de eficacia y toxicidad.[
Fase de posconsolidación
La terapia de posconsolidación está diseñada para tratar la posible ERM después de un TCMH.[
Evidencia (todos los tratamientos):
- En un estudio aleatorizado se comparó el tratamiento con dosis altas y trasplante autógeno de médula ósea (TAMO) con células purificadas con 3 ciclos de quimioterapia de consolidación intensiva. Además, después de finalizar la quimioterapia o el TAMO, se asignó al azar a los pacientes para interrumpir el tratamiento o recibir isotretinoína durante 6 meses. Los resultados de SSC y SG descritos a continuación reflejan el desenlace desde el momento de cada aleatorización.[
2424 ]; [3030 ][Nivel de evidencia A1]- La tasa de SSC a 5 años fue significativamente mejor en el grupo del TAMO (30 %) que en el grupo de quimioterapia de consolidación (19 %, P = 0,04). No hubo ninguna diferencia significativa en las tasas de SG a 5 años entre los 2 grupos (39 vs. 30 %, P = 0,08).
- Los pacientes que recibieron isotretinoína tuvieron una tasa de SSC a 5 años más alta que los pacientes que no recibieron tratamiento de mantenimiento (42 vs. 31 %), aunque la diferencia no fue significativa (P = 0,12).
- La tasa de SG fue más alta en los pacientes asignados al azar a recibir isotretinoína (50 %) que en los pacientes que interrumpieron el tratamiento (39 %), pero esta diferencia no fue significativa (P = 0,10).
- En un ensayo retrospectivo no aleatorizado de una sola institución, se compararon a los pacientes que recibieron terapia con GM-CSF y anticuerpo 3F8 anti-GD2 después de someterse a un TCMH autógeno o quimioterapia convencional.[
4747 ] El grupo de pacientes era una mezcla de aquellos derivados para el tratamiento inicial o para terapias adicionales, e incluyó a pacientes con enfermedad resistente al tratamiento o en recaída, algunos de los cuales ya se habían sometido a TCMH autógeno en las instituciones remitentes. En el grupo de TCMH autógeno, el tiempo desde la primera quimioterapia o desde el TCMH autógeno hasta el comienzo de la terapia con GM-CSF y anticuerpo 3F8 anti-GD2 fue significativamente más prolongado. El grupo de TCMH autógeno también tuvo a un número mayor de pacientes con riesgo muy alto.- Se observó una tendencia hacia una mejor SSC para los pacientes del grupo de terapia con GM-CSF, anticuerpo 3F8 anti-GD2 y TCMH autógeno (65 vs. 51 %, P = 0,128), pero no hubo diferencia estadísticamente significativa en la SG entre el grupo de quimioterapia sola y el grupo de TCMH autógeno.
- En un ensayo de fase III del COG (ANBL0032 [NCT00026312]), los participantes que se habían sometido antes a un TCMH fueron asignados al azar a recibir dinutuximab administrado con GM-CSF e IL-2 junto con isotretinoína, versus isotretinoína sola.[
4545 ]- El uso de inmunoterapia e isotretinoína (tasa de SSC, 66 %) fue superior al tratamiento estándar de mantenimiento con isotretinoína (tasa de SSC, 46 %). Como resultado, la inmunoterapia administrada después de un TCMH se considera el estándar de atención en los ensayos del COG para la enfermedad de riesgo alto.
- A partir de los estudios del COG, la Administración de Alimentos y Medicamentos (FDA) de los Estados Unidos aprobó el dinutuximab.
- Se dispuso de un seguimiento a largo plazo (mediana de seguimiento, 9,97 años; intervalo, 0,7–15,3 años) de 226 pacientes aptos. La tasa de SSC a 5 años fue del 56,6 % (± 4,7 %) para los pacientes asignados al azar a recibir inmunoterapia (n = 114) versus el 46,1 % (± 5,1 %) para los asignados al azar a recibir isotretinoína sola (n = 112) (P = 0,042). La tasa de SC a 5 años fue del 73,2 % (± 4,2 %) en los pacientes que recibieron inmunoterapia, versus 56,6 % (± 5,1 %) en los pacientes que recibieron isotretinoína (P = 0,045). De los 122 pacientes, 13 que recibieron dinutuximab presentaron anticuerpos antiquiméricos humanos (HACA). Las concentraciones plasmáticas de dinutuximab, HACA y receptor α de la IL-2 soluble no se correlacionaron con la SSC, la SG ni con efectos tóxicos clínicamente significativos.[
4848 ][Nivel de evidencia B1] - Una vez interrumpida la aleatorización para el ensayo ANBL0032, todos los pacientes fueron asignados a recibir inmunoterapia. Ya que se disponía de datos de seguimiento más prolongados para 1183 pacientes, se observó que los resultados de supervivencia y toxicidad fueron similares a los de informes anteriores. Para los pacientes mayores de 18 meses en el momento del diagnóstico con enfermedad en estadio 4 del INSS (n = 662), la tasa de SSC a 5 años fue del 57 %, y la tasa de SG fue del 70,9 %. Los efectos tóxicos fueron similares a los notificados para la cohorte aleatorizada. Entre los pacientes con datos disponibles, las concentraciones más altas de dinutuximab y el genotipo del receptor Fc 3A γ (FCGR3A) se relacionaron con una SSC superior.[
4949 ] - Para tratar a los pacientes con neuroblastoma, es frecuente el uso de los anticuerpos anti-GD2 junto con la modulación del sistema inmunitario a fin de mejorar la actividad antineoplásica del anticuerpo. La eficacia clínica de uno de estos anticuerpos condujo a que la FDA aprobara el dinutuximab. Es posible que la respuesta del paciente a la inmunoterapia obedezca en parte a una variación del funcionamiento inmunitario entre pacientes. Un anticuerpo anti-GD2, llamado 3F8, de uso exclusivo para el tratamiento del neuroblastoma en una institución, le indica a los linfocitos citolíticos naturales que destruyan las células de neuroblastoma. Sin embargo, a veces, la interacción de antígenos HLA y los subtipos de receptores de inmunoglobulina de los linfocitos citolíticos naturales (KIR) inhiben la acción de los linfocitos citolíticos naturales.[
5050 ,5151 ] Este hallazgo se confirmó y se amplió mediante un análisis de desenlaces de pacientes del estudio nacional aleatorizado COG-ANBL0032 (NCT00026312) tratados con dinutuximab (anticuerpo anti-GD2) combinado con GM-CSF e IL-2. En el estudio, se encontró que ciertos genotipos positivos para KIR y el ligando de KIR se relacionaban con mejores desenlaces en pacientes tratados con inmunoterapia.[5252 ][Nivel de evidencia A2] La presencia de ligandos inhibitorios de KIR o del ligando de KIR se vinculó con una disminución del efecto de la inmunoterapia. Por lo tanto, los genes del sistema inmunitario del paciente ayudan a determinar la respuesta del neuroblastoma a la inmunoterapia. Son necesarios más estudios para determinar si esta forma de genotipificación del sistema inmunitario puede guiar la selección de pacientes que deben recibir ciertos tipos de inmunoterapias.
- En un estudio europeo se comparó el uso de dinutuximab-beta (dinutuximab producido en células de hámster en lugar de células de ratones) y el uso de dinutuximab beta con IL-2 en inyecciones subcutáneas (SC) como terapia de mantenimiento después de dosis altas de quimioterapia y TCMH autógeno. Además, todos los pacientes recibieron isotretinoína.[
5353 ]- La adición de IL-2 SC no mejoró el desenlace; la tasa de SSC a 3 años fue del 56 % para los pacientes tratados con dinutuximab-beta y del 60 % para los pacientes tratados con dinutuximab-beta e IL-2 SC (P = 0,76).
- Tampoco hubo diferencias en la incidencia de recaída/progresión ni en la SG a 5 años.
- Los pacientes tratados con IL-2 presentaron tasas más elevadas de fiebre, dolor, reacción alérgica, síndrome de fuga capilar, neurotoxicidad y toxicidad gastrointestinal. En este estudio, solo el 62 % de los pacientes asignados al azar al grupo de IL-2 recibieron la terapia planificada debido a la toxicidad.
- En un segundo ensayo de la SIOPEN se informó lo siguiente:[
5353 ]- Las tasas de respuesta y las tasas de SSC y SG a 2 años no difirieron en los pacientes tratados con IL-2 versus los tratados sin IL-2.
- Posteriormente, la SIOPEN eliminó la IL-2 del tratamiento de posconsolidación estándar.
- En un tercer ensayo aleatorizado de fase II de la SIOPEN se comparó el tratamiento con IL-2 con el tratamiento sin IL-2.[
5454 ]- Las tasas de respuesta, SSC y SG no fueron significativamente diferentes con IL-2 o sin esta, pero la toxicidad fue mayor en el grupo con IL-2.
A partir de los datos de la SIOPEN, el COG retiró la IL-2 de la inmunoterapia de posconsolidación estándar.
La terapia con MIBG radiactiva se ha usado para tratar el neuroblastoma recidivante con cierto éxito. Esta terapia ha demostrado ser segura y viable para incorporarla al régimen de tratamiento de los niños con neuroblastoma de riesgo alto recién diagnosticado.[
En un ensayo clínico multinstitucional de fase II de niños con neuroblastoma de riesgo alto, se evaluaron 2 años de la terapia de continuación con eflornitina (conocida antes como difluorometilornitina [DFMO]), un inhibidor oral de la ornitina descarboxilasa.[
Otro abordaje que se ha estudiado para pacientes en primera remisión tras la finalización del tratamiento estándar es la administración de una vacuna de gangliósidos GD2/GD3. En un ensayo aleatorizado, que incluyó sobre todo a pacientes en primera remisión, la introducción temprana del betaglucano junto con una vacuna GD2/GD3 aumentó los valores de anticuerpos GD2/GD3 sin aumentar los efectos tóxicos. Las tasas de SSP fueron similares para los pacientes de ambos grupos de tratamiento aleatorizados. No obstante, los pacientes con valores más altos tuvieron tasas de SSP más favorables, con independencia del grupo de tratamiento.[
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el
Ensayos clínicos en curso
Realizar una
Referencias:
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Cotterill SJ, Pearson AD, Pritchard J, et al.: Late relapse and prognosis for neuroblastoma patients surviving 5 years or more: a report from the European Neuroblastoma Study Group "Survey". Med Pediatr Oncol 36 (1): 235-8, 2001.
- Mertens AC, Yasui Y, Neglia JP, et al.: Late mortality experience in five-year survivors of childhood and adolescent cancer: the Childhood Cancer Survivor Study. J Clin Oncol 19 (13): 3163-72, 2001.
- Morgenstern DA, London WB, Stephens D, et al.: Metastatic neuroblastoma confined to distant lymph nodes (stage 4N) predicts outcome in patients with stage 4 disease: A study from the International Neuroblastoma Risk Group Database. J Clin Oncol 32 (12): 1228-35, 2014.
- Kushner BH, LaQuaglia MP, Cardenas FI, et al.: Stage 4N neuroblastoma before and during the era of anti-GD2 immunotherapy. Int J Cancer 153 (12): 2019-2031, 2023.
- Kushner BH, LaQuaglia MP, Bonilla MA, et al.: Highly effective induction therapy for stage 4 neuroblastoma in children over 1 year of age. J Clin Oncol 12 (12): 2607-13, 1994.
- Park JR, Scott JR, Stewart CF, et al.: Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high-risk neuroblastoma: a Children's Oncology Group study. J Clin Oncol 29 (33): 4351-7, 2011.
- Decarolis B, Schneider C, Hero B, et al.: Iodine-123 metaiodobenzylguanidine scintigraphy scoring allows prediction of outcome in patients with stage 4 neuroblastoma: results of the Cologne interscore comparison study. J Clin Oncol 31 (7): 944-51, 2013.
- Yanik GA, Parisi MT, Shulkin BL, et al.: Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54 (4): 541-8, 2013.
- Pinto N, Naranjo A, Hibbitts E, et al.: Predictors of differential response to induction therapy in high-risk neuroblastoma: A report from the Children's Oncology Group (COG). Eur J Cancer 112: 66-79, 2019.
- Furman WL, McCarville B, Shulkin BL, et al.: Improved Outcome in Children With Newly Diagnosed High-Risk Neuroblastoma Treated With Chemoimmunotherapy: Updated Results of a Phase II Study Using hu14.18K322A. J Clin Oncol 40 (4): 335-344, 2022.
- Berthold F, Faldum A, Ernst A, et al.: Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: results of the randomized open-label GPOH trial NB2004-HR. Ann Oncol 31 (3): 422-429, 2020.
- Garaventa A, Poetschger U, Valteau-Couanet D, et al.: Randomized Trial of Two Induction Therapy Regimens for High-Risk Neuroblastoma: HR-NBL1.5 International Society of Pediatric Oncology European Neuroblastoma Group Study. J Clin Oncol 39 (23): 2552-2563, 2021.
- De Ioris MA, Crocoli A, Contoli B, et al.: Local control in metastatic neuroblastoma in children over 1 year of age. BMC Cancer 15: 79, 2015.
- von Allmen D, Davidoff AM, London WB, et al.: Impact of Extent of Resection on Local Control and Survival in Patients From the COG A3973 Study With High-Risk Neuroblastoma. J Clin Oncol 35 (2): 208-216, 2017.
- Englum BR, Rialon KL, Speicher PJ, et al.: Value of surgical resection in children with high-risk neuroblastoma. Pediatr Blood Cancer 62 (9): 1529-35, 2015.
- DeCou JM, Bowman LC, Rao BN, et al.: Infants with metastatic neuroblastoma have improved survival with resection of the primary tumor. J Pediatr Surg 30 (7): 937-40; discussion 940-1, 1995.
- Castel V, Tovar JA, Costa E, et al.: The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37 (11): 1574-8, 2002.
- Simon T, Häberle B, Hero B, et al.: Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31 (6): 752-8, 2013.
- Adkins ES, Sawin R, Gerbing RB, et al.: Efficacy of complete resection for high-risk neuroblastoma: a Children's Cancer Group study. J Pediatr Surg 39 (6): 931-6, 2004.
- Seemann NM, Erker C, Irwin MS, et al.: Survival effect of complete surgical resection of the primary tumor in patients with metastatic, high-risk neuroblastoma in a large Canadian cohort. Pediatr Blood Cancer 70 (6): e30286, 2023.
- Holmes K, Pötschger U, Pearson ADJ, et al.: Influence of Surgical Excision on the Survival of Patients With Stage 4 High-Risk Neuroblastoma: A Report From the HR-NBL1/SIOPEN Study. J Clin Oncol 38 (25): 2902-2915, 2020.
- Desai AV, Applebaum MA, Karrison TG, et al.: Efficacy of post-induction therapy for high-risk neuroblastoma patients with end-induction residual disease. Cancer 128 (15): 2967-2977, 2022.
- Matthay KK, Villablanca JG, Seeger RC, et al.: Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med 341 (16): 1165-73, 1999.
- Berthold F, Boos J, Burdach S, et al.: Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. Lancet Oncol 6 (9): 649-58, 2005.
- Pritchard J, Cotterill SJ, Germond SM, et al.: High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatr Blood Cancer 44 (4): 348-57, 2005.
- Elborai Y, Hafez H, Moussa EA, et al.: Comparison of toxicity following different conditioning regimens (busulfan/melphalan and carboplatin/etoposide/melphalan) for advanced stage neuroblastoma: Experience of two transplant centers. Pediatr Transplant 20 (2): 284-9, 2016.
- Ladenstein R, Pötschger U, Pearson ADJ, et al.: Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol 18 (4): 500-514, 2017.
- Park JR, Kreissman SG, London WB, et al.: Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA 322 (8): 746-755, 2019.
- Matthay KK, Reynolds CP, Seeger RC, et al.: Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children's oncology group study. J Clin Oncol 27 (7): 1007-13, 2009.
- Yalçin B, Kremer LC, Caron HN, et al.: High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst Rev 8: CD006301, 2013.
- Hale GA, Arora M, Ahn KW, et al.: Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience. Bone Marrow Transplant 48 (8): 1056-64, 2013.
- Kreissman SG, Seeger RC, Matthay KK, et al.: Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol 14 (10): 999-1008, 2013.
- Haas-Kogan DA, Swift PS, Selch M, et al.: Impact of radiotherapy for high-risk neuroblastoma: a Children's Cancer Group study. Int J Radiat Oncol Biol Phys 56 (1): 28-39, 2003.
- Casey DL, Kushner BH, Cheung NK, et al.: Local Control With 21-Gy Radiation Therapy for High-Risk Neuroblastoma. Int J Radiat Oncol Biol Phys 96 (2): 393-400, 2016.
- Gatcombe HG, Marcus RB, Katzenstein HM, et al.: Excellent local control from radiation therapy for high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 74 (5): 1549-54, 2009.
- Liu KX, Naranjo A, Zhang FF, et al.: Prospective Evaluation of Radiation Dose Escalation in Patients With High-Risk Neuroblastoma and Gross Residual Disease After Surgery: A Report From the Children's Oncology Group ANBL0532 Study. J Clin Oncol 38 (24): 2741-2752, 2020.
- Casey DL, Kushner BH, Cheung NV, et al.: Dose-escalation is needed for gross disease in high-risk neuroblastoma. Pediatr Blood Cancer 65 (7): e27009, 2018.
- Braunstein SE, London WB, Kreissman SG, et al.: Role of the extent of prophylactic regional lymph node radiotherapy on survival in high-risk neuroblastoma: A report from the COG A3973 study. Pediatr Blood Cancer 66 (7): e27736, 2019.
- Polishchuk AL, Li R, Hill-Kayser C, et al.: Likelihood of bone recurrence in prior sites of metastasis in patients with high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 89 (4): 839-45, 2014.
- Li R, Polishchuk A, DuBois S, et al.: Patterns of Relapse in High-Risk Neuroblastoma Patients Treated With and Without Total Body Irradiation. Int J Radiat Oncol Biol Phys 97 (2): 270-277, 2017.
- Mazloom A, Louis CU, Nuchtern J, et al.: Radiation therapy to the primary and postinduction chemotherapy MIBG-avid sites in high-risk neuroblastoma. Int J Radiat Oncol Biol Phys 90 (4): 858-62, 2014.
- Casey DL, Pitter KL, Kushner BH, et al.: Radiation Therapy to Sites of Metastatic Disease as Part of Consolidation in High-Risk Neuroblastoma: Can Long-term Control Be Achieved? Int J Radiat Oncol Biol Phys 100 (5): 1204-1209, 2018.
- Hattangadi JA, Rombi B, Yock TI, et al.: Proton radiotherapy for high-risk pediatric neuroblastoma: early outcomes and dose comparison. Int J Radiat Oncol Biol Phys 83 (3): 1015-22, 2012.
- Yu AL, Gilman AL, Ozkaynak MF, et al.: Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363 (14): 1324-34, 2010.
- Cheung NK, Cheung IY, Kushner BH, et al.: Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol 30 (26): 3264-70, 2012.
- Kushner BH, Ostrovnaya I, Cheung IY, et al.: Lack of survival advantage with autologous stem-cell transplantation in high-risk neuroblastoma consolidated by anti-GD2 immunotherapy and isotretinoin. Oncotarget 7 (4): 4155-66, 2016.
- Yu AL, Gilman AL, Ozkaynak MF, et al.: Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin Cancer Res 27 (8): 2179-2189, 2021.
- Desai AV, Gilman AL, Ozkaynak MF, et al.: Outcomes Following GD2-Directed Postconsolidation Therapy for Neuroblastoma After Cessation of Random Assignment on ANBL0032: A Report From the Children's Oncology Group. J Clin Oncol 40 (35): 4107-4118, 2022.
- Forlenza CJ, Boudreau JE, Zheng J, et al.: KIR3DL1 Allelic Polymorphism and HLA-B Epitopes Modulate Response to Anti-GD2 Monoclonal Antibody in Patients With Neuroblastoma. J Clin Oncol 34 (21): 2443-51, 2016.
- Venstrom JM, Zheng J, Noor N, et al.: KIR and HLA genotypes are associated with disease progression and survival following autologous hematopoietic stem cell transplantation for high-risk neuroblastoma. Clin Cancer Res 15 (23): 7330-4, 2009.
- Erbe AK, Wang W, Carmichael L, et al.: Neuroblastoma Patients' KIR and KIR-Ligand Genotypes Influence Clinical Outcome for Dinutuximab-based Immunotherapy: A Report from the Children's Oncology Group. Clin Cancer Res 24 (1): 189-196, 2018.
- Ladenstein R, Pötschger U, Valteau-Couanet D, et al.: Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 19 (12): 1617-1629, 2018.
- Ladenstein RL, Poetschger U, Valteau-Couanet D, et al.: Randomization of dose-reduced subcutaneous interleukin-2 (scIL2) in maintenance immunotherapy (IT) with anti-GD2 antibody dinutuximab beta (DB) long-term infusion (LTI) in front–line high-risk neuroblastoma patients: Early results from the HR-NBL1/SIOPEN trial. [Abstract] J Clin Oncol 37 (suppl 15): A-10013, 2019.
Also available onlineAlso available online . Last accessed August 21, 2023. - Weiss BD, Yanik G, Naranjo A, et al.: A safety and feasibility trial of 131 I-MIBG in newly diagnosed high-risk neuroblastoma: A Children's Oncology Group study. Pediatr Blood Cancer 68 (10): e29117, 2021.
- Sholler GLS, Ferguson W, Bergendahl G, et al.: Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci Rep 8 (1): 14445, 2018.
- Oesterheld J, Ferguson W, Kraveka JM, et al.: Eflornithine as Postimmunotherapy Maintenance in High-Risk Neuroblastoma: Externally Controlled, Propensity Score-Matched Survival Outcome Comparisons. J Clin Oncol 42 (1): 90-102, 2024.
- Cheung IY, Mauguen A, Modak S, et al.: Effect of Oral β-Glucan on Antibody Response to Ganglioside Vaccine in Patients With High-Risk Neuroblastoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 9 (2): 242-250, 2023.
Tratamiento del neuroblastoma en estadio 4S del INSS y estadio MS del INRG
Los pacientes en estadio 4S del International Neuroblastoma Staging System (INSS) son menores de 12 meses y tienen un tumor primario en estadio 1 o estadio 2 del INSS, mientras que los pacientes en estadio MS del International Neuroblastoma Risk Group (INRG) son menores de 18 meses y tienen un tumor primario de cualquier estadio. Ambos sistemas de estratificación usan la misma definición de patrón limitado de metástasis.
La decisión del INRG Task Force de reemplazar la categoría de enfermedad 4S con la de la nueva definición de MS se tomó a partir de los informes de desenlaces favorables en un número pequeño de lactantes con tumores primarios L2 y patrones metastásicos, incluso en pacientes de 12 a 18 meses de vida.[
Los lactantes con enfermedad en estadio MS del INRG tienen características biológicas más favorables y resultados superiores, a pesar de recibir una terapia menos intensiva. La tasa de supervivencia sin complicaciones (SSC) a 5 años fue del 86 % y la tasa de supervivencia general (SG) fue del 95 %. Para los pacientes con tumores con amplificación de MYCN, la tasa de SSC a los 5 años fue del 60 % y la tasa de SG fue del 65 %.[
Muchos pacientes con neuroblastoma en estadio 4S o MS no necesitan tratamiento. No obstante, los tumores con características biológicas desfavorables o los pacientes sintomáticos por hepatomegalia progresiva y compromiso orgánico tienen un mayor riesgo de muerte y se tratan con dosis bajas o moderadas de quimioterapia. Entre un 8 % y un 10 % de estos pacientes tendrán amplificación de MYCN y se tratan con protocolos de riesgo alto.[
Para obtener más información sobre el esquema de clasificación para el neuroblastoma en estadio 4S o MS del Children's Oncology Group (COG), consultar el
Opciones de tratamiento del neuroblastoma en estadio 4S o MS
No existe ningún abordaje estándar para el tratamiento del neuroblastoma en estadio 4S o MS.
Las opciones de tratamiento del neuroblastoma en estadio 4S o MS son las siguientes:
-
Observación con cuidados médicos de apoyoObservación con cuidados médicos de apoyo (para pacientes asintomáticos con características biológicas tumorales favorables). -
QuimioterapiaQuimioterapia (para pacientes sintomáticos o con características biológicas desfavorables). -
RadioterapiaRadioterapia (rara vez para pacientes con síntomas relacionados con hepatomegalia por enfermedad metastásica).
La resección del tumor primario no se relaciona con un desenlace mejor.[
Observación con cuidados médicos de apoyo
La observación con cuidados médicos de apoyo se usa para tratar a pacientes asintomáticos con tumores de características biológicas favorables.
El tratamiento de los niños con enfermedad en estadio 4S o MS depende del cuadro clínico inicial.[
Quimioterapia
La quimioterapia se usa para tratar a pacientes sintomáticos o pacientes con características biológicas desfavorables. Los pacientes con indicios de crecimiento tumoral rápido en las primeras semanas de vida necesitan una intervención inmediata con quimioterapia para evitar el síndrome compartimental abdominal potencialmente irreversible e insuficiencia renal y hepática.[
Los lactantes diagnosticados con un neuroblastoma en estadio 4S o MS del INSS, en particular aquellos con hepatomegalia o aquellos menores de 2 meses con características de riesgo alto o hepatomegalia, quizás presenten un deterioro clínico rápido y se pueden beneficiar de la iniciación temprana del tratamiento.[
Para evaluar mejor a este grupo de pacientes en estadio 4S, se formuló un sistema de puntaje para medir los signos y síntomas de deterioro o compromiso.[
Se han utilizado distintos regímenes de quimioterapia (ciclofosfamida sola, carboplatino y etopósido, o ciclofosfamida, doxorrubicina y vincristina) para tratar a los pacientes sintomáticos. El abordaje consiste en administrar quimioterapia sola mientras los síntomas persisten con el fin de evitar los efectos tóxicos, que contribuyen a una supervivencia más precaria. Además, a menudo se recomienda el uso de dosis más bajas de quimioterapia para los lactantes muy pequeños o de bajo peso, junto con factores estimulantes de colonias de granulocitos después de cada ciclo de quimioterapia.
Evidencia (quimioterapia para la enfermedad en estadio 4S o MS):
- En el estudio del COG ANBL0531 (NCT00499616) se estudió en forma prospectiva a un subconjunto de pacientes en estadio 4S que tenían tumores sin amplificación de MYCN con disfunción orgánica inminente o características biológicas desfavorables (tipo histológico desfavorable o índice de DNA diploide). Se inscribió a 49 pacientes, 41 sintomáticos y 28 con características biológicas desfavorables. Se asignó a los pacientes a recibir 2, 4 u 8 ciclos de quimioterapia a partir de las características biológicas del tumor, la edad del paciente y los síntomas.[
99 ][Nivel de evidencia C1]- La tasa de SG a 3 años fue del 81,4 %.
- De las 9 muertes que ocurrieron, 8 fueron de pacientes menores de 2 meses de vida en el momento del diagnóstico. Las causas de 5 muertes fueron complicaciones agudas de una hepatomegalia de progresión rápida (es decir, síndrome compartimental abdominal, insuficiencia renal, insuficiencia respiratoria, coagulopatía e infección). Los pacientes menores de 40 días de vida en el momento del diagnóstico tuvieron un riesgo de muerte 13 veces mayor, en comparación con los pacientes mayores de 47 días de vida. El estudio se modificó después de las primeras 5 muertes, y se ordenó la administración inmediata de quimioterapia para los pacientes con enfermedad en estadio 4S menores de 2 meses de vida en el momento del diagnóstico y con hepatomegalia progresiva. No hubo muertes por complicaciones de la hepatomegalia en los lactantes inscritos después, incluso 18 lactantes con menos de 2 meses de vida.
La descompresión abdominal quirúrgica de urgencia se puede usar para evitar el deterioro respiratorio y mejorar la ventilación.[
1212 ,1313 ] - En este estudio se confirmó el desenlace precario de los pacientes con características biológicas desfavorables (ID = 1, anomalías cromosómicas segmentarias [pérdida de heterocigosis 1p o 11p, características histológicas desfavorables] sin amplificación de MYCN), en comparación con los pacientes sintomáticos con características biológicas favorables.
- Los pacientes con muerte tardía fallecieron debido a enfermedad metastásica y tenían características biológicas desfavorables.
- Se inscribieron 80 pacientes con enfermedad en estadio 4S en el ensayo COG-P9641. De los pacientes, 41 presentaban neuroblastoma asintomático en estadio 4S y fueron tratados con cirugía o biopsia solas, y 39 recibieron tratamiento con cirugía y quimioterapia.[
1414 ]- En general, la tasa de SSC a 5 años fue del 77 %, y la tasa de SG fue del 91 %.
- La tasa de SSC a 5 años fue del 63 % para los pacientes tratados con cirugía o biopsia sola y del 95 % para los pacientes tratados con cirugía y quimioterapia (P = 0,0016).
- La tasa de SG a 5 años fue del 84 % para los pacientes tratados con cirugía o biopsia sola y del 97 % para los pacientes tratados con cirugía y quimioterapia (P = 0,1302).
Antes se pensaba que la toxicidad de la quimioterapia era responsable de la supervivencia más precaria de los pacientes con enfermedad en estadio 4S; sin embargo, el uso de quimioterapia en el ensayo COG-P9641 se limitó a situaciones clínicas específicas con un número recomendado de ciclos.
- Asimismo, en el ensayo COG-P9641, los lactantes asintomáticos con enfermedad en estadio 4S del INSS y características biológicas favorables (sin amplificación de MYCN) no recibieron quimioterapia hasta que presentaron enfermedad progresiva o síntomas clínicos.[
1414 ]- Los niños que se tornaron sintomáticos presentaban insuficiencia orgánica relacionada con la enfermedad y complicaciones infecciosas que llevaron a una SG inferior en comparación con los que recibieron quimioterapia inmediata (4-8 ciclos de tratamiento). La tasa de SG a 3 años para los lactantes que no recibieron quimioterapia fue del 84 % versus el 97 % de los lactantes que recibieron quimioterapia (P = 0,1321).
- En el ensayo COG-ANBL0531, el tratamiento se asignó según los síntomas, la edad y las características biológicas del tumor.[
99 ]- La tasa de SG a 2 años para pacientes en estadio 4S del INSS fue del 81 %, que es inferior a la notificada en otros ensayos cooperativos, como el COG-P9641.
- Muchos pacientes inscritos en el estudio ANBL0531 estaban más enfermos que los pacientes que ingresaron en estudios anteriores, en parte porque no se requirió de la biopsia del tumor en lactantes sintomáticos. Los ensayos anteriores incluyeron principalmente a pacientes asintomáticos y la mayoría tenía características biológicas favorables.
- Se realizó un estudio prospectivo de 125 lactantes con tumores en estadio 4S sin amplificación de MYCN, tumores primarios en estadio 3 del INSS y centellografía ósea positiva sin cambios en el hueso cortical documentados en radiografías simples o tomografía computarizada.[
1111 ] Se utilizó un puntaje de los síntomas antes del tratamiento para determinar el tratamiento inicial; se recomendó observación para los lactantes con puntajes bajos de síntomas (n = 86) y quimioterapia para los lactantes con puntajes altos de síntomas (n = 37).La quimioterapia para los pacientes con puntajes altos de síntomas incluyó de 2 a 4 cursos de 3 días de carboplatino y etopósido; si los síntomas persistían o la enfermedad progresaba, se administraban hasta 4 cursos de 5 días de ciclofosfamida, doxorrubicina y vincristina. La mitad de los pacientes se sometieron a resección parcial o completa del tumor primario.
- No hubo diferencias en las SSC y SG a 2 años entre los pacientes sintomáticos y asintomáticos (tasa de SSC, 87 vs. 88 %; tasa de SG, 98 vs. 97 %), aunque muchos de los investigadores prefirieron administrar quimioterapia en presencia de un puntaje bajo de síntomas.
- Para los lactantes con puntajes bajos de síntomas, no hubo ninguna diferencia en el resultado de los lactantes no tratados inicialmente (n = 56; tasa de SG, 93 %) y los lactantes tratados (n = 30; tasa de SG, 86 %).
- La tasa de SG fue del 90 % para los lactantes con puntajes altos de síntomas.
- No hubo ninguna diferencia significativa en las tasas de SG a 2 años entre los pacientes con tumores primarios irresecables y los pacientes con tumores primarios resecables (97 vs. 100 %), ni entre los pacientes con centellografía ósea negativa y positiva, sin anomalías radiológicas (100 vs. 97 %).
Radioterapia (para los pacientes con síntomas relacionados con hepatomegalia por enfermedad metastásica)
En casos poco frecuentes de hepatomegalia marcada en lactantes sintomáticos con neuroblastoma en estadio MS (4S) que no respondieron a la quimioterapia, se administraron dosis muy bajas de radioterapia. En una serie de 41 lactantes sintomáticos con enfermedad en estadio MS, se administró radioterapia a 5 lactantes y 3 de ellos murieron.[
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el
Ensayos clínicos en curso
Realizar una
Referencias:
- Monclair T, Brodeur GM, Ambros PF, et al.: The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27 (2): 298-303, 2009.
- Taggart DR, London WB, Schmidt ML, et al.: Prognostic value of the stage 4S metastatic pattern and tumor biology in patients with metastatic neuroblastoma diagnosed between birth and 18 months of age. J Clin Oncol 29 (33): 4358-64, 2011.
- Irwin MS, Naranjo A, Zhang FF, et al.: Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol 39 (29): 3229-3241, 2021.
- Canete A, Gerrard M, Rubie H, et al.: Poor survival for infants with MYCN-amplified metastatic neuroblastoma despite intensified treatment: the International Society of Paediatric Oncology European Neuroblastoma Experience. J Clin Oncol 27 (7): 1014-9, 2009.
- Guglielmi M, De Bernardi B, Rizzo A, et al.: Resection of primary tumor at diagnosis in stage IV-S neuroblastoma: does it affect the clinical course? J Clin Oncol 14 (5): 1537-44, 1996.
- Katzenstein HM, Bowman LC, Brodeur GM, et al.: Prognostic significance of age, MYCN oncogene amplification, tumor cell ploidy, and histology in 110 infants with stage D(S) neuroblastoma: the pediatric oncology group experience--a pediatric oncology group study. J Clin Oncol 16 (6): 2007-17, 1998.
- Nickerson HJ, Matthay KK, Seeger RC, et al.: Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children's Cancer Group study. J Clin Oncol 18 (3): 477-86, 2000.
- Steele M, Jones NL, Ng V, et al.: Successful liver transplantation in an infant with stage 4S(M) neuroblastoma. Pediatr Blood Cancer 60 (3): 515-7, 2013.
- Twist CJ, Naranjo A, Schmidt ML, et al.: Defining Risk Factors for Chemotherapeutic Intervention in Infants With Stage 4S Neuroblastoma: A Report From Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (2): 115-124, 2019.
- Hsu LL, Evans AE, D'Angio GJ: Hepatomegaly in neuroblastoma stage 4s: criteria for treatment of the vulnerable neonate. Med Pediatr Oncol 27 (6): 521-8, 1996.
- De Bernardi B, Gerrard M, Boni L, et al.: Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol 27 (7): 1034-40, 2009.
- Keene DJ, Minford J, Craigie RJ, et al.: Laparostomy closure in stage 4S neuroblastoma. J Pediatr Surg 46 (1): e1-4, 2011.
- Harper L, Perel Y, Lavrand F, et al.: Surgical management of neuroblastoma-related hepatomegaly: do material and method really count? Pediatr Hematol Oncol 25 (4): 313-7, 2008.
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.
Tratamiento de neuroblastoma recidivante
El crecimiento del tumor resultante de la maduración del niño se deberá diferenciar de la progresión tumoral mediante biopsia y revisión de las características histológicas. Los pacientes a veces tienen una enfermedad en maduración persistente con captación de metayodobencilguanidina (MlBG) que no afecta el desenlace; en especial, en pacientes con enfermedad de riesgo bajo e intermedio.[
A veces se identifican variantes subclonales de ALK u otras lesiones en la vía MAPK en el momento del diagnóstico, con una expansión clonal subsiguiente en el momento de la recaída. En consecuencia, un muestreo en serie de tumores progresivos quizás lleve a la identificación de variantes aprovechables terapéuticamente.[
La secuenciación de tumores de neuroblastoma recurrentes y resistentes al tratamiento de pacientes pediátricos (n = 59) y adultos jóvenes (n = 1) incluidos en el ensayo NCI-COG Pediatric MATCH reveló alteraciones genómicas que se consideraron aprovechable para el tratamiento en los grupos del estudio MATCH en 27 de 60 tumores (45 %).[
Si un niño presenta un neuroblastoma recidivante que en un principio se diagnosticó como enfermedad de riesgo alto, por lo general, el pronóstico es precario a pesar del tratamiento intensivo adicional.[
Factores pronósticos del neuroblastoma recidivante
Se realizó un análisis exhaustivo de los patrones de recaída utilizando la base de datos del International Neuroblastoma Risk Group (INRG) en pacientes diagnosticados o inscritos entre 1989 y 2017.[
- En 1833 niños, el patrón de la primera recaída incluyó sitio local aislado (19 %), sitio solo a distancia (65 %) y sitios combinados (16 %).
- Los pacientes con fracaso local aislado tenían características pronósticas más favorables.
- Los pacientes con enfermedad en estadio 3 tenían más probabilidades de sufrir un fracaso local aislado que los pacientes en todos los demás estadios (49 vs. 16 %, P < 0,001).
- Las tasas de supervivencia general (SG) a 5 años difirieron significativamente según el patrón de recaída, con una tasa del 64 % para sitio local aislado, del 23 % para sitio solo a distancia y del 26 % para sitios combinados (P < 0,001).
- Después de realizar ajustes por edad, estadio y estado de MYCN, los pacientes con fracaso local aislado (cociente de riesgos instantáneos [CRI] ajustado, 0,46; P < 0,001) y fracaso solo a distancia (CRI ajustado, 0,57; P < 0,001) siguieron teniendo un menor riesgo de muerte, en comparación con los pacientes con fracaso combinado.
Se usó la base de datos del INRG para examinar las características clínicas y biológicas que son pronósticas de la supervivencia tras la recaída o progresión del neuroblastoma con el patrón estadístico MS del INRG Staging System (INRGSS). De los 1511 pacientes diagnosticados entre 1984 y 2021 que cumplían los criterios de elegibilidad, se identificaron 209 pacientes con recaída. Los criterios de elegibilidad incluían pacientes menores de 365 días en el momento del diagnóstico inicial con enfermedad en estadio MS del INRGSS o en estadio 4S del INSS, o pacientes de 365 a 546 días con enfermedad en estadio 4 del INSS y metástasis limitada al hígado, la piel o la médula ósea.[
- En este grupo, la mediana de tiempo hasta la primera recaída fue de 8,16 meses.
- La mayoría de las recaídas tenían un componente de fracaso metastásico. Estas metástasis se produjeron con mayor frecuencia en sitios fuera del hígado, la piel y la médula ósea.
- La tasa de SG a 5 años fue del 62 % en los pacientes tratados a partir de 2001.
En el International Neuroblastoma Risk Group Project se realizó un análisis del árbol de supervivencia de las características clínicas y biológicas (definidas en el momento del diagnóstico) relacionadas con la supervivencia después de la recaída en 2266 pacientes de neuroblastoma participantes en grandes ensayos clínicos de grupos de ensayos clínicos bien establecidos de todo el mundo.[
- La tasa de SG en toda la población con recaída fue del 20 %.
- Entre los pacientes en todos los estadios de enfermedad en el momento del diagnóstico, la amplificación de MYCN predijo un pronóstico más precario, medido como SG a 5 años.
- Entre los pacientes con diagnóstico de enfermedad en estadio 4 del International Neuroblastoma Staging System (INSS) sin amplificación, edad mayor de 18 meses y concentración alta de lactato–deshidrogenasa (LDH) predijeron un pronóstico precario.
- Entre los pacientes con amplificación de MYCN, aquellos con diagnóstico de enfermedad en estadios 1 y 2 tuvieron un mejor pronóstico que los de enfermedad en estadios 3 y 4.
- Entre los pacientes con tumores sin amplificación de MYCN que no están en estadio 4, los pacientes menores de 18 meses con hiperdiploidía tuvieron un mejor pronóstico que los pacientes menores de 18 meses con diploidía; mientras que, entre los mayores de 18 meses, los pacientes con tumores diferenciados tuvieron un pronóstico mucho mejor que los pacientes con tumores indiferenciados y poco diferenciados.
Los factores pronósticos significativos para la supervivencia posterior a la recaída determinados en el momento del diagnóstico son los siguientes:[
- Edad.
- Estadio del INSS.
- Estado de MYCN.
- Tiempo desde el diagnóstico hasta la primera recaída.
- Concentración de LDH, ploidía y grado histológico de diferenciación tumoral (en menor medida).
La experiencia del Children's Oncology Group (COG) indica que la recuperación es posible en la mayoría de los pacientes con neuroblastomas de riesgo bajo y riesgo intermedio que presentan recidiva. El COG notificó tasas a 3 años de supervivencia sin complicaciones (SSC)del 88 % y de SG del 96 %, en pacientes de riesgo intermedio y tasas a 5 años de SSC del 89 % y de SG del 97 % en pacientes de riesgo bajo.[
Aunque, por lo general, la SG después de una recidiva en los niños que presentan inicialmente neuroblastoma de riesgo alto es muy precaria, en un estudio de una sola institución se halló una tasa de SG a 5 años del 35 % en los pacientes de neuroblastoma de riesgo alto en el momento de la primera recaída después de una remisión completa o enfermedad residual mínima (ERM) en quienes la recaída ocurrió en un solo sitio con una masa de tejido blando (algunos niños también tenían enfermedad en la médula ósea o en huesos en el momento de la recaída). Se sometió a todos los pacientes a resección quirúrgica de la enfermedad en el tejido blando. La amplificación de MYCN y la enfermedad de tejido blando multifocal se relacionaron con una supervivencia posterior a la progresión más precaria.[
En el
| Asignación al grupo de riesgo del COG | Opciones de tratamiento |
|---|---|
| COG = Children's Oncology Group; 131I-MIBG = yodo I 131- metayodobencilguanidina. | |
| Recidiva locorregional en pacientes clasificados inicialmente como de riesgo bajo | |
| |
|
| Recidiva metastásica en pacientes clasificados inicialmente como de riesgo bajo | |
| |
|
| |
|
| Recidiva locorregional en pacientes clasificados inicialmente como de riesgo intermedio | |
| |
|
| |
|
| Recidiva metastásica en pacientes clasificados inicialmente como de riesgo intermedio | |
| Recidiva en pacientes clasificados inicialmente como de riesgo alto | |
| |
|
| |
|
| |
|
| |
|
| Recidiva en el sistema nervioso central | |
| |
|
| |
|
Neuroblastoma recidivante en pacientes clasificados inicialmente como de riesgo bajo
Recidiva locorregional
Las opciones de tratamiento del neuroblastoma locorregional recidivante inicialmente clasificado como de riesgo bajo son las siguientes:
- Cirugía seguida de observación o quimioterapia.
- Quimioterapia y posible cirugía posterior.
El cáncer recidivante local o regional se extirpa cuando es posible.
Los pacientes con características biológicas favorables y recidiva regional más de 3 meses después de finalizar el tratamiento planificado se someten a observación si la resección de la recidiva es total o subtotal (≥90 % de resección). Aquellos con características biológicas favorables y un grado de resección más bajo se tratan con quimioterapia.[
Los lactantes menores de 1 año en el momento de la recidiva locorregional cuyos tumores tienen propiedades biológicas desfavorables se observan si la resección es total o subtotal. Si el grado de resección es más bajo que un grado subtotal, estos lactantes se tratan con quimioterapia. La quimioterapia se compone de dosis moderadas de carboplatino, ciclofosfamida, doxorrubicina y etopósido, o de ciclofosfamida y topotecán. La dosis acumulada de cada fármaco se mantiene baja para reducir al mínimo los efectos a largo plazo, como se usó en los ensayos previos del COG (COG-P9641 y COG-A3961).[
Evidencia (cirugía, seguida de observación o quimioterapia):
- En un estudio del COG sobre el tratamiento de pacientes de riesgo bajo con neuroblastomas en estadios 1, 2A, 2B y 4S, participaron 915 pacientes, de los cuales 800 eran asintomáticos, que se trataron con cirugía sola seguida de observación. Los demás pacientes recibieron quimioterapia con cirugía o sin esta.[
1717 ]- Alrededor del 10 % de los pacientes presentaron tumores progresivos o recidivantes.
- En el estudio, la mayoría de las recidivas se trataron con cirugía sola o quimioterapia en dosis moderadas, con cirugía o sin esta;
- y la mayoría de los pacientes se recuperaron de la enfermedad, como lo demuestran las tasas de SSC (89 %) y de SG (97 %) a 5 años.
Recidiva metastásica o enfermedad resistente al tratamiento estándar
Las opciones de tratamiento del neuroblastoma recidivante metastásico inicialmente clasificado como de riesgo bajo son las siguientes:
- Observación.
- Quimioterapia (en función de la edad del paciente, las características biológicas del tumor y el tratamiento previo; el tratamiento puede incluir terapias de riesgo intermedio o alto, según se hayan utilizado en el diagnóstico inicial).
- Cirugía seguida de quimioterapia.
El neuroblastoma recidivante metastásico o progresivo en un lactante clasificado inicialmente como de riesgo bajo y menor de 1 año en el momento de la recidiva se puede tratar de acuerdo con las características biológicas del tumor según se definió en los ensayos previos del COG (COG-P9641 y COG-A3961):
- Si las características biológicas son completamente favorables, la metástasis tiene un patrón 4S y la recidiva o progresión se presenta dentro de los 3 meses del diagnóstico, el paciente se observa y se atienden los síntomas según aparezcan.
- Si la progresión o la recidiva metastásica se produce más de 3 meses después del diagnóstico o no tiene un patrón 4S, el tumor primario se reseca, si es posible, y se administra quimioterapia.
La quimioterapia se compone de dosis moderadas de carboplatino, ciclofosfamida, doxorrubicina y etopósido. La dosis acumulada de cada fármaco se mantiene baja para reducir al mínimo los efectos a largo plazo, como se usó en ensayos previos del COG (COG-P9641 y COG-A3961).
Cualquier niño clasificado inicialmente como de riesgo bajo que tenga más de 18 meses en el momento de la recidiva metastásica o la enfermedad progresiva, y que no tenga una recidiva que siga el patrón del estadio 4S, por lo general, tiene un pronóstico precario y se trata de la siguiente forma:
- Tratamiento de riesgo alto.
Los pacientes con neuroblastoma recidivante metastásico se tratan como los pacientes con neuroblastoma de riesgo alto recién diagnosticado. Para obtener más información, consultar la sección
Neuroblastoma recidivante en pacientes clasificados inicialmente como de riesgo intermedio
En el estudio del COG ANBL0531 (NCT00499616) los pacientes con neuroblastoma de riesgo intermedio recién diagnosticado recibieron quimioterapia con carboplatino, etopósido, ciclofosfamida y doxorrubicina. En el protocolo se incluyó una terapia de recuperación para los pacientes que presentaron enfermedad progresiva no metastásica en los primeros 3 años desde la inscripción en el estudio. Se podía administrar a los pacientes hasta 6 ciclos de ciclofosfamida y topotecán. De los 29 pacientes que recibieron ciclofosfamida y topotecán, 18 no presentaron complicaciones, 9 tuvieron una recaída y 2 fallecieron. De los pacientes que experimentaron una respuesta inicial inadecuada a 8 ciclos de quimioterapia, 20 recibieron ciclofosfamida y topotecán. De esos 20 pacientes, 9 lograron una respuesta parcial muy buena o mejor; sin embargo, 6 pacientes presentaron enfermedad progresiva o recaída, y 1 falleció. Esto indica que se necesita una terapia más intensiva para los pacientes que no alcanzan el criterio de valoración definido para el tratamiento tras 8 ciclos de quimioterapia.[
En el estudio previo del COG para el neuroblastoma de riesgo intermedio (COG-A3961) se inscribió a 479 pacientes, de los cuales 42 presentaron enfermedad progresiva. La tasa de recidiva fue del 10 % para aquellos con características biológicas favorables y del 17 % para los que tienen características biológicas desfavorables. De los pacientes, 30 presentaron recidivas locorregionales, 11 recidivas metastásicas y 1 tenía ambos tipos de enfermedad recidivante. De los 42 pacientes, 6 murieron por la enfermedad y 36 respondieron al tratamiento. Por lo tanto, la mayoría de los pacientes con neuroblastoma de riesgo intermedio y progresión de la enfermedad se pueden recuperar.[
Recidiva locorregional
Las opciones de tratamiento del neuroblastoma con recidiva locorregional inicialmente clasificado como de riesgo intermedio son las siguientes:
- Cirugía (resección completa).
- Cirugía (resección incompleta) seguida de quimioterapia.
- Radioterapia. La radioterapia se considera solo para los pacientes con progresión de la enfermedad después de la quimioterapia y la revisión quirúrgica.[
1616 ]
La recidiva locorregional del neuroblastoma con características biológicas favorables que se presenta más de 3 meses después de terminar la quimioterapia, se puede tratar quirúrgicamente. Si la resección no alcanza a ser subtotal, es posible que se administre quimioterapia adicional. La quimioterapia debe seleccionarse en base a la quimioterapia previa recibida.[
Recidiva metastásica
La opción de tratamiento del neuroblastoma recidivante metastásico inicialmente clasificado como de riesgo intermedio es la siguiente:
- Tratamiento de riesgo alto.
Los pacientes con neuroblastoma recidivante metastásico se tratan como los pacientes con neuroblastoma de riesgo alto recién diagnosticado. Para obtener más información, consultar la sección
Neuroblastoma recidivante en pacientes clasificados inicialmente como de riesgo alto
Cualquier recidiva en pacientes clasificados inicialmente como de riesgo alto tiene un pronóstico muy precario.[
En un análisis de varios ensayos se incluyeron a 383 pacientes con neuroblastoma recidivante o progresivo que participaron en los ensayos de fase inicial de la era moderna del COG. La tasa de supervivencia sin progresión (SSP) a 1 año fue del 21 % y la tasa de SSP a 4 años fue del 6 %. Las tasas de SG fueron del 57 % a 1 año y del 20 % a los 4 años. Menos del 10 % de los pacientes no presentaron recidiva ni progresión posterior. La amplificación de MYCN permitió predecir peores tasas de SSP y SG.[
Las opciones de tratamiento del neuroblastoma recidivante o resistente al tratamiento en pacientes clasificados inicialmente como de riesgo alto son las siguientes:
- Quimioterapia en combinación con inmunoterapia.
- Temozolomida, irinotecán y dinutuximab.[
2121 ]
- Temozolomida, irinotecán y dinutuximab.[
- 131I-MIBG. 131I-MlBG solo, en combinación con otro tratamiento, o seguido de rescate de células madre.
- Tratamientos novedosos.
- Inhibidores de ALK para pacientes con variantes de ALK. En una serie de 20 pacientes con anomalías en ALK tratados con crizotinib, la tasa de respuesta fue del 15 %. De los pacientes, 2 tuvieron respuestas parciales y 1 tuvo una respuesta completa. Los 3 pacientes presentaban una variante somática ALK Arg1275Gln.[
2222 ][Nivel de evidencia C3] Lorlatinib ha mostrado actividad en pacientes con neuroblastoma recidivante con ALK anómalo. Las tasas de respuesta fueron del 13 % en pacientes menores de 18 años y del 47 % en pacientes de 18 años o más.[2323 ] Otros pacientes de cada cohorte obtuvieron respuestas menores, lo que se tradujo en tasas de respuesta modificadas del 30 % y el 67 %, respectivamente. En una serie de una sola institución se notificó que 9 de 13 pacientes adultos con neuroblastoma en recaída, que presentaba anomalías en ALK respondieron al lorlatinib.[2424 ] - Inhibidores de la EMA1. En un ensayo de fase II de adavosertib e irinotecán se informó que 3 de 20 pacientes con neuroblastoma recidivante tuvieron respuestas objetivas, lo que cumplió con el criterio principal de eficacia.[
2525 ]
- Inhibidores de ALK para pacientes con variantes de ALK. En una serie de 20 pacientes con anomalías en ALK tratados con crizotinib, la tasa de respuesta fue del 15 %. De los pacientes, 2 tuvieron respuestas parciales y 1 tuvo una respuesta completa. Los 3 pacientes presentaban una variante somática ALK Arg1275Gln.[
- Quimioterapia (estudios de fase I o II).
- Topotecán en combinación con ciclofosfamida o etopósido.[
2626 ] - Temozolomida con irinotecán.
- Topotecán en combinación con ciclofosfamida o etopósido.[
- Inmunoterapia. Se han evaluado nuevos fármacos anti-GD2 en pacientes con neuroblastoma recidivante o resistente al tratamiento. El anti-GD2 Hu14.18 se unió de forma química a la IL-2 y se combinó con el factor estimulante de colonias de granulocitos y macrófagos (GM-CSF), y en un ensayo de fase II de este régimen se notificaron pocas respuestas duraderas.[
2727 ]
La quimioterapia en combinación con inmunoterapia produce la mejor tasa de respuesta y duración de respuesta de los tratamientos para pacientes de riesgo alto con progresión de la enfermedad.
Evidencia (quimioterapia en combinación con inmunoterapia):
- El ensayo ANBL1221 (NCT01767194) fue el primer ensayo multicéntrico en el que se evaluó la terapia anti-GD2 combinada con quimioterapia en una cohorte de pacientes con neuroblastoma en recaída o resistente al tratamiento. Los pacientes en su primera recaída o progresión se asignaron de manera aleatoria para recibir irinotecán, temozolomida, dinutuximab y GM-CSF (I/T/DIN/GM-CSF), o temozolomida, irinotecán y temsirólimus.[
2121 ]; [2828 ][Nivel de evidencia C3]- De los 17 pacientes tratados con la combinación que incluía dinutuximab, 9 pacientes (53 %) tuvieron respuestas objetivas, en comparación con 1 de 18 pacientes tratados con el régimen que contenía temsirólimus.
- En una cohorte de ampliación, 36 pacientes adicionales se asignaron de manera no aleatoria para recibir I/T/DIN/GM-CSF; se observaron respuestas objetivas en 13 pacientes (36,1 %). En los 53 pacientes inscritos en el estudio y tratados con I/T/DIN/GM-CSF hubo 22 respuestas objetivas (41,5 %).[
2828 ][Nivel de evidencia C3] Este desenlace supera a cualquier otro publicado para los pacientes con neuroblastoma de riesgo alto resistente al tratamiento o en recaída.
- En un estudio retrospectivo de cohortes de 146 pacientes con neuroblastoma de riesgo alto que recibieron quimioinmunoterapia con I/T/DIN/GM-CSF en la primera recaída, se notificaron los siguientes resultados:[
2929 ][Nivel de evidencia C2]- El 49 % de los pacientes presentaron una respuesta objetiva, similar a la tasa de respuesta observada en el ensayo ANBL1221.
- De los pacientes con enfermedad estable o mejor en la primera evaluación de la enfermedad tras la quimioinmunoterapia, el 22 % presentó una respuesta mejorada (según los International Neuroblastoma Response Criteria) en la evaluación posterior. Solo el 13 % de los pacientes con enfermedad estable en la primera evaluación de la enfermedad presentaron eventualmente una respuesta objetiva, mientras que alrededor del 40 % de los pacientes con un estado inicial de respuesta mínima o parcial lograron una respuesta completa tras los ciclos posteriores. Los pacientes que recibieron más de 6 ciclos de tratamiento y continuaron con enfermedad estable tenían pocas probabilidades de alcanzar una respuesta objetiva.
- La mediana de SSP fue de 13,1 meses desde el inicio del tratamiento. La tasa de SSP a 1 año fue del 50 % y la tasa de SG a 2 años fue del 28 %.
- La mediana de duración de la respuesta fue de 15,9 meses.
- La mediana de SSP tras la interrupción de todo el tratamiento anticanceroso, incluso I/T/DIN/GM-CSF, fue de 10,4 meses.
Evidencia (131I-MIBG solo o en combinación con otro tratamiento):
- Para los niños con neuroblastoma recidivante o resistente al tratamiento, 131I-MlBG es una sustancia paliativa eficaz y se debe considerar sola o en combinación con quimioterapia (con rescate de células madre) en un ensayo clínico de investigación.[
3030 ,3131 ,3232 ,3333 ,3434 ,3535 ]; [3636 ,3737 ][Nivel de evidencia C1] - En un estudio retrospectivo realizado en América del Norte de más de 200 pacientes tratados con 131I-MIBG, se comparó a niños que tenían enfermedad recidivante o progresiva con niños que tenían enfermedad estable o persistente desde el momento del diagnóstico.[
3838 ]- La tasa de progresión inmediata después de la terapia con 131I-MIBG fue más baja y la tasa de SG a 2 años fue mejor (65 vs. 39 %) en los pacientes con enfermedad estable o persistente.
- Se notificó de modo retrospectivo sobre 8 pacientes sometidos a consolidación en tándem con 131I-MIBG, vincristina e irinotecán con trasplante de células madre hematopoyéticas (TCMH) autógeno seguido de busulfano y melfalán con TCMH autógeno;[
3737 ]- Se obtuvieron 3 respuestas completas, 2 respuestas parciales y 1 respuesta menor.
- En otros pacientes, se estudió el TCMH autógeno solo con aumento gradual de la dosis de 131I-MIBG y carboplatino, etopósido y melfalán.[
3939 ]- Después de la quimioterapia de inducción, se trató a 27 pacientes con resistencia al tratamiento y 15 pacientes con enfermedad progresiva; se obtuvieron 4 respuestas. Se trató a 8 pacientes que exhibieron una respuesta parcial a la inducción y se obtuvieron 3 respuestas.
- La incidencia del 12 % de síndrome de obstrucción sinusoidal fue un factor limitante de la dosis.
- En un ensayo aleatorizado de fase II se incluyó a 105 pacientes evaluables que recibieron tratamiento con 131I-MIBG sola, 131I-MIBG con irinotecán y vincristina, o 131I-MIBG con vorinostat.[
4040 ]- Los pacientes incluidos en el grupo de vorinostat tuvieron la tasa de respuesta más alta (32 %).
- Los pacientes tratados con MIBG sola o con irinotecán/vincristina tuvieron tasas de respuesta del 14 %.
- En un ensayo de fase II de un solo grupo se incluyeron 30 pacientes con neuroblastoma en recaída o resistente al tratamiento que se trataron con 131I-MIBG y topotecán.[
4141 ]- Se notificó una tasa de respuestas del 13%.
Evidencia (quimioterapia):
- En un estudio, la combinación de irinotecán y temozolomida tuvo una tasa de respuesta del 15 %.[
4242 ][Nivel de evidencia B4] - En un estudio retrospectivo, se notificó sobre 74 pacientes que recibieron 92 ciclos de ifosfamida, carboplatino y etopósido; este número incluía a 37 pacientes que recibieron rescate de células madre de sangre periférica después de responder a esta combinación de fármacos.[
4343 ]- Se lograron regresiones de la enfermedad (respuestas mayores y menores) en 14 de 17 pacientes (82 %) con una recaída nueva, 13 de 26 pacientes (50 %) con neuroblastoma resistente al tratamiento, y 12 de 34 pacientes (35 %) tratados por enfermedad progresiva durante la quimioterapia (P = 0,005).
- Los efectos tóxicos de grado 3 fueron poco frecuentes.
- El topotecán, en combinación con ciclofosfamida sola o con etopósido, se ha utilizado en pacientes con enfermedad recidivante que no recibieron inicialmente topotecán. Las tasas de respuesta fueron del 32 % (18 de 57) para los pacientes que recibieron topotecán y ciclofosfamida y del 19 % (11 de 59) para los pacientes que recibieron topotecán solo.[
2626 ][Nivel de evidencia A2] - Se han empleado dosis altas de carboplatino, irinotecán o temozolomida para tratar a pacientes con enfermedad resistente al tratamiento o nuevas recaídas (tras un tratamiento que incluía topotecán) que se producen después del tratamiento (tasa de respuesta objetiva, 68 %). Sin embargo, este régimen no se usa para tratar a pacientes cuya enfermedad progresa mientras reciben el tratamiento.[
4444 ]
Tradicionalmente, el trasplante alogénico tiene una tasa de éxito baja en los neuroblastomas recidivantes o progresivos. En un estudio retrospectivo de registros, el TCMH alogénico luego de un TCMH autógeno previo tuvo beneficios mínimos. La recidiva de la enfermedad sigue siendo la causa más común de fracaso del tratamiento.[
Están en curso ensayos clínicos de vacunas diseñadas para inducir en el huésped la producción de anticuerpos antigangliósidos que pueden replicar la actividad antineoplásica de los anticuerpos monoclonales administrados por vía intravenosa. Los pacientes también se someten a un tratamiento con betaglucano, que tiene un espectro amplio de efectos inmunoestimulantes y tiene sinergia con los anticuerpos monoclonales anti-GD2/GD3. En un estudio de fase I de 15 niños con neuroblastoma de riesgo alto, el tratamiento se toleró sin ningún efecto tóxico que limitara la dosis.[
En un ensayo de fase I/II, el uso de células T con expresión del receptor de antígeno quimérico (CAR) anti GD2 fue viable e inocuo para el tratamiento de niños con neuroblastoma de riesgo alto en recaída o resistente al tratamiento. Con este tratamiento se obtuvo una tasa de respuesta del 63 %.[
Neuroblastoma recidivante en el sistema nervioso central
El compromiso del sistema nervioso central (SNC), aunque es poco frecuente durante el cuadro clínico inicial, se produce en el 3 % al 10 % de los pacientes de neuroblastoma recidivante. Las recaídas en el SNC representaron un 6 % de todas las recaídas metastásicas en una serie de 1161 primeras recaídas en 1977 pacientes con enfermedad en estadio 4 tratados en un ensayo de pacientes con neuroblastoma de riesgo alto.[
Los factores de riesgo importante de recaída en el SNC identificados en el ensayo International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN) fueron las características del paciente y de la enfermedad en el momento del diagnóstico. Estas características incluyeron el sexo femenino (cociente de riesgos instantáneos [CRI], 2,0; P = 0,016), la amplificación de MYCN (CRI, 2,4; P = 0,0008), la enfermedad hepática (CRI, 2,5; P = 0,01) o el compromiso de más de un sistema o compartimento metastásico (CRI, 7,1; P = 0,047). Ni la quimioterapia de dosis altas ni la inmunoterapia se relacionaron con un mayor riesgo de recidiva. A lo largo del tiempo, los investigadores observaron una incidencia estable de recaídas en el SNC.[
Las recaídas en el SNC son casi siempre mortales, con una mediana de tiempo hasta la muerte de 6 meses. Las tasas de SG a 1 y 3 años tras la recaída fueron del 25 % y el 7 %, respectivamente, en el ensayo SIOPEN.[
Las opciones de tratamiento del neuroblastoma recidivante en el SNC son las siguientes:
- Cirugía y radioterapia.
- Quimioterapia (incluidos los regímenes que contienen temozolomida) en combinación con cirugía y radioterapia.
- Abordajes terapéuticos nuevos.
Por lo general, los abordajes actuales de tratamiento incluyen la erradicación de la enfermedad residual macro y microscópica en el SNC y la enfermedad sistémica residual mínima precursora de recaídas futuras. Las intervenciones neuroquirúrgicas sirven para disminuir el edema, controlar la hemorragia y extirpar la masa tumoral antes de iniciar la radioterapia.
Una sola institución tuvo cierto éxito cuando probó la radioinmunoterapia compartimental intraventricular con administración intratecal de anticuerpos monoclonales anti-GD2 radioyodados, en combinación con dosis de 18 Gy o 21 Gy de radiación craneoespinal con refuerzos para la enfermedad macroscópica en el SNC en pacientes con neuroblastoma metastásico recidivante en el SNC.[
Quizás algunos de los pacientes con una supervivencia prolongada tras una recaída inicial en el SNC presenten una segunda recaída tras la irradiación craneoespinal (ICE). Los datos publicados sobre pacientes que presentan una segunda recaída en el SNC son limitados. Una segunda recaída en el SNC augura un pronóstico precario.[
En un estudio de una sola institución, que incluyó 128 pacientes tratados con CSI para la primera recaída en el SNC, 40 pacientes presentaron una segunda recaída en el SNC al cabo de una mediana de 6,3 meses desde el tratamiento inicial con ICE. Los desenlaces de los pacientes tras una segunda recaída en el SNC son precarios, aunque el tratamiento con radioterapia en el momento de la segunda recaída en el SNC puede relacionarse con una SG más prolongada.[
- La tasa de supervivencia a 1 año fue del 32,5 %.
- Los pacientes de este grupo con compromiso leptomeníngeo inicial tenían más probabilidades de recaer que los que presentaban solo lesiones parenquimatosas (CRI, 2,5; IC 95 %, 1,3–4,9; P = 0,006). La mediana de tiempo transcurrido hasta la segunda recaída en el SNC fue de 6,8 meses; el 51 % de las recaídas se produjeron fuera del campo de refuerzo de la ICE.
- La mayoría de los pacientes (24 de 40) recibieron radioterapia como parte del abordaje multimodal para la segunda recaída en el SNC. La administración de radioterapia en el momento de la segunda recaída en el SNC se relacionó con una SG mejor (mediana, 30 vs. 5,1 meses; CRI, 0,5; orden logarítmico P < 0,001).
- De los 40 pacientes, 8 recibieron radioinmunoterapia intratecal compartimental (RITc) con anticuerpos monoclonales anti-GD2 radioyodados como parte del tratamiento. La mediana de SG prolongada desde el momento de la segunda recaída fue de 22 meses para los pacientes que recibieron RITc. En comparación, la mediana de SG fue de 5 meses para los pacientes que no recibieron RITc. De estos pacientes, 5 habían recibido antes RITc en el momento de la recaída inicial en el SNC.
Opciones de tratamiento en evaluación clínica del neuroblastoma recidivante o resistente al tratamiento
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): en el NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), que se conoce como Pediatric MATCH, se emparejarán fármacos de terapia dirigida con cambios moleculares específicos identificados en el tumor (resistente al tratamiento o recidivante) de un paciente. Los niños y adolescentes de 1 a 21 años son aptos para participar en este ensayo.
Los pacientes que presentan tumores con variantes moleculares comprendidas en los grupos de tratamiento del Pediatric MATCH podrán inscribirse para recibir tratamiento en este ensayo. Para obtener más información en inglés, consultar el
portal de Internet del NCIportal de Internet del NCI y elportal de Internet ClinicalTrials.govportal de Internet ClinicalTrials.gov . - ADVL1621 (NCT02332668) (A Phase I/II Study of Pembrolizumab [MK-3475] in Children With Advanced Melanoma or a PD-L1–Positive Advanced, Relapsed or Refractory Solid Tumor or Lymphoma): en la parte 1 de este estudio se medirá la dosis máxima tolerada, se confirmará la dosis y se determinará la dosis recomendada de fase II para el tratamiento con pembrolizumab. En la parte 2 del estudio se evaluará en más profundidad la inocuidad y la eficacia de la dosis pediátrica recomendada para la fase II.
Ensayos clínicos en curso
Realizar una
Referencias:
- Marachelian A, Shimada H, Sano H, et al.: The significance of serial histopathology in a residual mass for outcome of intermediate risk stage 3 neuroblastoma. Pediatr Blood Cancer 58 (5): 675-81, 2012.
- Taggart DR, Han MM, Quach A, et al.: Comparison of iodine-123 metaiodobenzylguanidine (MIBG) scan and [18F]fluorodeoxyglucose positron emission tomography to evaluate response after iodine-131 MIBG therapy for relapsed neuroblastoma. J Clin Oncol 27 (32): 5343-9, 2009.
- Schleiermacher G, Javanmardi N, Bernard V, et al.: Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 32 (25): 2727-34, 2014.
- Padovan-Merhar OM, Raman P, Ostrovnaya I, et al.: Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PLoS Genet 12 (12): e1006501, 2016.
- Eleveld TF, Oldridge DA, Bernard V, et al.: Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47 (8): 864-71, 2015.
- Schramm A, Köster J, Assenov Y, et al.: Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47 (8): 872-7, 2015.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- London WB, Castel V, Monclair T, et al.: Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol 29 (24): 3286-92, 2011.
- Pole JG, Casper J, Elfenbein G, et al.: High-dose chemoradiotherapy supported by marrow infusions for advanced neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 9 (1): 152-8, 1991.
- Castel V, Cañete A, Melero C, et al.: Results of the cooperative protocol (N-III-95) for metastatic relapses and refractory neuroblastoma. Med Pediatr Oncol 35 (6): 724-6, 2000.
- Lau L, Tai D, Weitzman S, et al.: Factors influencing survival in children with recurrent neuroblastoma. J Pediatr Hematol Oncol 26 (4): 227-32, 2004.
- Saylors RL, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001.
- Kramer K, Kushner BH, Modak S, et al.: Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol 97 (3): 409-18, 2010.
- Vo KT, DuBois SG, Neuhaus J, et al.: Pattern and predictors of sites of relapse in neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 69 (9): e29616, 2022.
- Campbell K, Kao PC, Naranjo A, et al.: Clinical and biological features prognostic of survival after relapse or progression of INRGSS stage MS pattern neuroblastoma: A report from the International Neuroblastoma Risk Group (INRG) project. Pediatr Blood Cancer 70 (2): e30054, 2023.
- Baker DL, Schmidt ML, Cohn SL, et al.: Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363 (14): 1313-23, 2010.
- Strother DR, London WB, Schmidt ML, et al.: Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30 (15): 1842-8, 2012.
- Murphy JM, Lim II, Farber BA, et al.: Salvage rates after progression of high-risk neuroblastoma with a soft tissue mass. J Pediatr Surg 51 (2): 285-8, 2016.
- Twist CJ, Schmidt ML, Naranjo A, et al.: Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children's Oncology Group Study ANBL0531. J Clin Oncol 37 (34): 3243-3255, 2019.
- London WB, Bagatell R, Weigel BJ, et al.: Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children's Oncology Group early-phase trials. Cancer 123 (24): 4914-4923, 2017.
- Mody R, Naranjo A, Van Ryn C, et al.: Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol 18 (7): 946-957, 2017.
- Foster JH, Voss SD, Hall DC, et al.: Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children's Oncology Group Study (ADVL0912). Clin Cancer Res 27 (13): 3543-3548, 2021.
- Goldsmith KC, Park JR, Kayser K, et al.: Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med 29 (5): 1092-1102, 2023.
- Stiefel J, Kushner BH, Roberts SS, et al.: Anaplastic Lymphoma Kinase Inhibitors for Therapy of Neuroblastoma in Adults. JCO Precis Oncol 7: e2300138, 2023.
- Cole KA, Ijaz H, Surrey LF, et al.: Pediatric phase 2 trial of a WEE1 inhibitor, adavosertib (AZD1775), and irinotecan for relapsed neuroblastoma, medulloblastoma, and rhabdomyosarcoma. Cancer 129 (14): 2245-2255, 2023.
- London WB, Frantz CN, Campbell LA, et al.: Phase II randomized comparison of topotecan plus cyclophosphamide versus topotecan alone in children with recurrent or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol 28 (24): 3808-15, 2010.
- Shusterman S, Naranjo A, Van Ryn C, et al.: Antitumor Activity and Tolerability of hu14.18-IL2 with GMCSF and Isotretinoin in Recurrent or Refractory Neuroblastoma: A Children's Oncology Group Phase II Study. Clin Cancer Res 25 (20): 6044-6051, 2019.
- Mody R, Yu AL, Naranjo A, et al.: Irinotecan, Temozolomide, and Dinutuximab With GM-CSF in Children With Refractory or Relapsed Neuroblastoma: A Report From the Children's Oncology Group. J Clin Oncol 38 (19): 2160-2169, 2020.
- Lerman BJ, Li Y, Carlowicz C, et al.: Progression-Free Survival and Patterns of Response in Patients With Relapsed High-Risk Neuroblastoma Treated With Irinotecan/Temozolomide/Dinutuximab/Granulocyte-Macrophage Colony-Stimulating Factor. J Clin Oncol 41 (3): 508-516, 2023.
- DuBois SG, Groshen S, Park JR, et al.: Phase I Study of Vorinostat as a Radiation Sensitizer with 131I-Metaiodobenzylguanidine (131I-MIBG) for Patients with Relapsed or Refractory Neuroblastoma. Clin Cancer Res 21 (12): 2715-21, 2015.
- Polishchuk AL, Dubois SG, Haas-Kogan D, et al.: Response, survival, and toxicity after iodine-131-metaiodobenzylguanidine therapy for neuroblastoma in preadolescents, adolescents, and adults. Cancer 117 (18): 4286-93, 2011.
- Matthay KK, Yanik G, Messina J, et al.: Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol 25 (9): 1054-60, 2007.
- Matthay KK, Tan JC, Villablanca JG, et al.: Phase I dose escalation of iodine-131-metaiodobenzylguanidine with myeloablative chemotherapy and autologous stem-cell transplantation in refractory neuroblastoma: a new approaches to Neuroblastoma Therapy Consortium Study. J Clin Oncol 24 (3): 500-6, 2006.
- Matthay KK, Quach A, Huberty J, et al.: Iodine-131--metaiodobenzylguanidine double infusion with autologous stem-cell rescue for neuroblastoma: a new approaches to neuroblastoma therapy phase I study. J Clin Oncol 27 (7): 1020-5, 2009.
- DuBois SG, Chesler L, Groshen S, et al.: Phase I study of vincristine, irinotecan, and ¹³¹I-metaiodobenzylguanidine for patients with relapsed or refractory neuroblastoma: a new approaches to neuroblastoma therapy trial. Clin Cancer Res 18 (9): 2679-86, 2012.
- Johnson K, McGlynn B, Saggio J, et al.: Safety and efficacy of tandem 131I-metaiodobenzylguanidine infusions in relapsed/refractory neuroblastoma. Pediatr Blood Cancer 57 (7): 1124-9, 2011.
- French S, DuBois SG, Horn B, et al.: 131I-MIBG followed by consolidation with busulfan, melphalan and autologous stem cell transplantation for refractory neuroblastoma. Pediatr Blood Cancer 60 (5): 879-84, 2013.
- Zhou MJ, Doral MY, DuBois SG, et al.: Different outcomes for relapsed versus refractory neuroblastoma after therapy with (131)I-metaiodobenzylguanidine ((131)I-MIBG). Eur J Cancer 51 (16): 2465-72, 2015.
- Yanik GA, Villablanca JG, Maris JM, et al.: 131I-metaiodobenzylguanidine with intensive chemotherapy and autologous stem cell transplantation for high-risk neuroblastoma. A new approaches to neuroblastoma therapy (NANT) phase II study. Biol Blood Marrow Transplant 21 (4): 673-81, 2015.
- DuBois SG, Granger MM, Groshen S, et al.: Randomized Phase II Trial of MIBG Versus MIBG, Vincristine, and Irinotecan Versus MIBG and Vorinostat for Patients With Relapsed or Refractory Neuroblastoma: A Report From NANT Consortium. J Clin Oncol 39 (31): 3506-3514, 2021.
- Sevrin F, Kolesnikov-Gauthier H, Cougnenc O, et al.: Phase II study of 131 I-metaiodobenzylguanidine with 5 days of topotecan for refractory or relapsed neuroblastoma: Results of the French study MIITOP. Pediatr Blood Cancer 70 (11): e30615, 2023.
- Bagatell R, London WB, Wagner LM, et al.: Phase II study of irinotecan and temozolomide in children with relapsed or refractory neuroblastoma: a Children's Oncology Group study. J Clin Oncol 29 (2): 208-13, 2011.
- Kushner BH, Modak S, Kramer K, et al.: Ifosfamide, carboplatin, and etoposide for neuroblastoma: a high-dose salvage regimen and review of the literature. Cancer 119 (3): 665-71, 2013.
- Kushner BH, Kramer K, Modak S, et al.: Differential impact of high-dose cyclophosphamide, topotecan, and vincristine in clinical subsets of patients with chemoresistant neuroblastoma. Cancer 116 (12): 3054-60, 2010.
- Hale GA, Arora M, Ahn KW, et al.: Allogeneic hematopoietic cell transplantation for neuroblastoma: the CIBMTR experience. Bone Marrow Transplant 48 (8): 1056-64, 2013.
- Flaadt T, Ladenstein RL, Ebinger M, et al.: Anti-GD2 Antibody Dinutuximab Beta and Low-Dose Interleukin 2 After Haploidentical Stem-Cell Transplantation in Patients With Relapsed Neuroblastoma: A Multicenter, Phase I/II Trial. J Clin Oncol 41 (17): 3135-3148, 2023.
- Kushner BH, Cheung IY, Modak S, et al.: Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res 20 (5): 1375-82, 2014.
- Kushner BH, Ostrovnaya I, Cheung IY, et al.: Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-GD2 immunotherapy and isotretinoin: a prospective Phase II study. Oncoimmunology 4 (7): e1016704, 2015.
- Del Bufalo F, De Angelis B, Caruana I, et al.: GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N Engl J Med 388 (14): 1284-1295, 2023.
- Berlanga P, Pasqualini C, Pötschger U, et al.: Central nervous system relapse in high-risk stage 4 neuroblastoma: The HR-NBL1/SIOPEN trial experience. Eur J Cancer 144: 1-8, 2021.
- Kramer K, Kushner B, Heller G, et al.: Neuroblastoma metastatic to the central nervous system. The Memorial Sloan-kettering Cancer Center Experience and A Literature Review. Cancer 91 (8): 1510-9, 2001.
- Matthay KK, Brisse H, Couanet D, et al.: Central nervous system metastases in neuroblastoma: radiologic, clinical, and biologic features in 23 patients. Cancer 98 (1): 155-65, 2003.
- Luo LY, Kramer K, Cheung NV, et al.: Reduced-dose craniospinal irradiation for central nervous system relapsed neuroblastoma. Pediatr Blood Cancer 67 (9): e28364, 2020.
- Tringale KR, Wolden SL, Casey DL, et al.: Clinical outcomes of pediatric patients receiving multimodality treatment of second central nervous system relapse of neuroblastoma. Pediatr Blood Cancer 70 (2): e30075, 2023.
Actualizaciones más recientes a este resumen (05 / 23 / 2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se incorporaron cambios editoriales en este resumen.
El
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del neuroblastoma. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del neuroblastoma son:
- Steven DuBois, MD, MS (Dana Farber Cancer Institute)
- Andrea A. Hayes-Dixon, MD, FACS, FAAP (Howard University)
- Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children's Cancer Center and Blood Disorders Harvard Medical School)
- Nita Louise Seibel, MD (National Cancer Institute)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento del neuroblastoma. Bethesda, MD: National Cancer Institute. Actualización: <MM/DD/YYYY>. Disponible en:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del
Última revisión: 2024-05-23
Esta información no reemplaza el consejo de un médico. Ignite Healthwise, LLC, niega toda garantía y responsabilidad por el uso de esta información. El uso que usted haga de esta información implica que usted acepta los
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
Page Footer
Quiero...
Audiencia
Sitios seguros para miembros
Información sobre The Cigna Group
Aviso legal
Los planes individuales y familiares de seguro médico y dental están asegurados por Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc. y Cigna HealthCare of Texas, Inc. Los planes de beneficios de salud y de seguro de salud de grupo están asegurados o administrados por CHLIC, Connecticut General Life Insurance Company (CGLIC) o sus afiliadas (puedes ver
Todas las pólizas de seguros y los planes de beneficios de grupo contienen exclusiones y limitaciones. Para conocer la disponibilidad, los costos y detalles completos de la cobertura, comunícate con un agente autorizado o con un representante de ventas de Cigna. Este sitio web no está dirigido a los residentes de New Mexico.