Childhood Acute Lymphoblastic Leukemia Treatment (PDQ®): Treatment - Health Professional Information [NCI]
General Information About Childhood Acute Lymphoblastic Leukemia (ALL)
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer, including ALL, has been slowly increasing since 1975.[
Incidence
ALL, the most common cancer diagnosed in children, represents approximately 25% of cancer diagnoses among children younger than 15 years.[
A sharp peak in ALL incidence is observed among children aged 1 to 4 years (81 cases per 1 million per year), with rates decreasing to 24 cases per 1 million by age 10 years.[
The incidence of ALL appears to be highest in American Indian or Alaska Native children and adolescents (65.9 cases per 1 million) and Hispanic children and adolescents (48 cases per 1 million).[
Anatomy
Childhood ALL originates in the T and B lymphoblasts in tissues with hematopoietic progenitor cells, such as the bone marrow and thymus (see Figure 1). 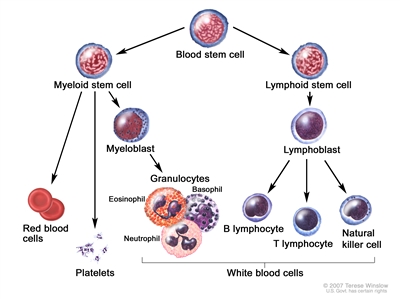
Figure 1. Blood cell development. Different blood and immune cell lineages, including T and B lymphocytes, differentiate from a common blood stem cell.
Marrow involvement of acute leukemia as seen by light microscopy is defined as follows:
- M1: Fewer than 5% blast cells.
- M2: 5% to 25% blast cells.
- M3: Greater than 25% blast cells.
Almost all patients with ALL present with an M3 marrow.
Morphology
In the past, ALL lymphoblasts were classified using the French-American-British (FAB) criteria as having L1, L2, or L3 morphology.[
Most cases of ALL that show L3 morphology express surface immunoglobulin (Ig) and have a MYC gene translocation identical to those seen in Burkitt lymphoma (i.e., t(8;14)(q24;q32), t(2;8)) that join MYC to one of the Ig genes. Patients with this specific rare form of leukemia (mature B-cell or Burkitt leukemia) should be treated according to protocols for Burkitt lymphoma. For more information about the treatment of mature B-cell lymphoma/leukemia and Burkitt lymphoma/leukemia, see Childhood Non-Hodgkin Lymphoma Treatment. Rarely, blasts with L1/L2 (not L3) morphology will express surface Ig.[
Risk Factors for Developing ALL
The primary accepted risk factors for ALL and associated genes (when relevant) include the following:
- Prenatal exposure to x-rays.
- Postnatal exposure to high doses of radiation (e.g., therapeutic radiation previously used for conditions such as tinea capitis and thymus enlargement).
- Previous treatment with chemotherapy.
- Genetic conditions that include the following:
- Down syndrome. For more information, see the Down syndrome section.
- Neurofibromatosis (NF1).[
14 ] - Bloom syndrome (BLM).[
15 ] - Fanconi anemia (multiple genes; ALL is observed much less frequently than acute myeloid leukemia [AML]).[
16 ] - Ataxia telangiectasia (ATM).[
17 ] - Li-Fraumeni syndrome (TP53).[
18 ,19 ,20 ] - Constitutional mismatch repair deficiency (biallelic variant of MLH1, MSH2, MSH6, and PMS2).[
21 ,22 ]
- Low- and high-penetrance inherited genetic variants.[
23 ] For more information, see the Low- and high-penetrance inherited genetic variants section. - Carriers of a constitutional Robertsonian translocation that involves chromosomes 15 and 21 and carriers of constitutional ring chromosome 21 are specifically and highly predisposed to developing intrachromosomal amplification of chromosome 21 (iAMP21) ALL.[
24 ,25 ]
Down syndrome
Children with Down syndrome have an increased risk of developing both ALL and AML,[
A genome-wide association study found that four susceptibility loci associated with B-ALL in the non-Down syndrome population (IKZF1, CDKN2A, ARID5B, and GATA3) were also associated with susceptibility to ALL in children with Down syndrome.[
Approximately one-half to two-thirds of cases of acute leukemia in children with Down syndrome are ALL, and about 2% to 3% of childhood ALL cases occur in children with Down syndrome.[
Patients with ALL and Down syndrome have a lower incidence of both favorable (ETV6::RUNX1 fusion and hyperdiploidy [51–65 chromosomes]) and unfavorable (BCR::ABL1 or KMT2A::AFF1 fusions and hypodiploidy [<44 chromosomes]) genomic alterations and a near absence of T-cell phenotype.[
Approximately 50% to 60% of cases of ALL in children with Down syndrome have genomic alterations affecting CRLF2 that generally result in overexpression of the protein produced by this gene, which dimerizes with the interleukin-7 receptor alpha to form the receptor for the cytokine thymic stromal lymphopoietin.[
Based on the relatively small number of published series, it does not appear that genomic CRLF2 aberrations in patients with Down syndrome and ALL have prognostic relevance.[
Approximately 20% to 30% of ALL cases arising in children with Down syndrome have somatically acquired JAK1 or JAK2 variants,[
IKZF1 gene deletions, observed in 20% to 35% of patients with Down syndrome and ALL, have been associated with a significantly worse outcome in this group of patients.[
Approximately 10% of patients with Down syndrome and ALL have genomic alterations leading to overexpression or abnormal activation of the CEBPD, CEBPA, and CEBPE genes.[
Low- and high-penetrance inherited genetic variants
Genetic predisposition to ALL can be divided into several broad categories, as follows:
- Association with genetic syndromes. Increased risk can be associated with the genetic syndromes listed above in which ALL is observed, although it is not the primary manifestation of the condition.
- Common alleles. Another category for genetic predisposition includes common alleles with relatively small effect sizes that are identified by genome-wide association studies. Genome-wide association studies have identified a number of germline (inherited) genetic polymorphisms that are associated with the development of childhood ALL.[
23 ] For example, the risk alleles of ARID5B are associated with the development of hyperdiploid (51–65 chromosomes) B-ALL. ARID5B is a gene that encodes a transcriptional factor important in embryonic development, cell type–specific gene expression, and cell growth regulation.[49 ,50 ] Other genes with polymorphisms associated with increased risk of ALL include GATA3,[51 ]IKZF1,[49 ,50 ,52 ]CDKN2A,[53 ]CDKN2B,[52 ,53 ]CEBPE,[49 ]PIP4K2A,[51 ,54 ] and TP63.[55 ]Genetic risk factors for T-ALL share some overlap with the genetic risk factors for B-ALL, but unique risk factors also exist. A genome-wide association study identified a risk allele near USP7 that was associated with an increased risk of developing T-ALL (odds ratio, 1.44) but not B-ALL. The risk allele was shown to be associated with reduced USP7 transcription, which is consistent with the finding that somatic loss-of-function variants in USP7 are observed in patients with T-ALL. USP7 germline and somatic variants are generally mutually exclusive and are most commonly observed in T-ALL patients with TAL1 alterations.[
56 ]Genetic risk factors that have similar impact for developing both B-ALL and T-ALL include CDKN2A, CDKN2B, and 8q24.21 (cis distal enhancer region variants for MYC).[
56 ] - Rare germline variants with high penetrance. Germline variants that cause pathogenic changes in genes associated with ALL and that are observed in kindreds with familial ALL (i.e., large effect sizes) comprise another category of genetic predisposition to ALL. Many of the genes associated with ALL risk play key roles in B-cell development (e.g., PAX5, ETV6, and IKZF1).[
57 ]- PAX5. A germline variant in PAX5 that substitutes serine for glycine at amino acid 183 and that reduces PAX5 activity has been identified in several families that experienced multiple cases of ALL.[
58 ,59 ] - ETV6. Several germline ETV6 variants that lead to loss of ETV6 function have been identified in kindreds affected by both thrombocytopenia and ALL.[
60 ,61 ,62 ,63 ,64 ] Sequencing of ETV6 in remission (i.e., germline) specimens identified variants that were potentially related to ALL in approximately 1% of children with ALL that were evaluated.[60 ] Most of the germline variants (approximately 75%) were shown to be deleterious for ETV6 function, and 70% of cases with a deleterious germline ETV6 variant had a hyperdiploid karyotype. The remaining cases with a deleterious variant had diploid ALL, with a transcriptional profile similar to that of cases with ETV6::RUNX1 fusion–positive ALL.[64 ] - TP53. Pathogenic germline TP53 variants are associated with an increased risk of ALL.[
65 ] A study of 3,801 children with ALL observed that 26 patients (0.7%) had a pathogenic TP53 germline variant, with an associated odds ratio of 5.2 for ALL development.[65 ] Compared with ALL in children with TP53 wild-type status or TP53 variants of unknown significance, ALL in children with pathogenic germline TP53 variants was associated with older age at diagnosis (15.5 years vs. 7.3 years), hypodiploidy (65% vs. 1%), inferior EFS and overall survival, and a higher risk of second cancers. - IKZF1. Germline IKZF1 variants were identified in a kindred with familial ALL and in 43 of 4,963 (0.9%) children with sporadic ALL. Most (22 of 28) IKZF1 variants were shown to adversely affect IKZF1 gene function.[
66 ] Germline variants in IKZF1 have been identified in hereditary hypogammaglobulinemia. In one series, 2 of 29 affected patients developed B-ALL during childhood.[67 ]
- PAX5. A germline variant in PAX5 that substitutes serine for glycine at amino acid 183 and that reduces PAX5 activity has been identified in several families that experienced multiple cases of ALL.[
Prenatal origin of childhood ALL
Development of ALL is a multistep process in most cases, with more than one genomic alteration required for frank leukemia to develop. In at least some cases of childhood ALL, the initial genomic alteration occurs in utero. Evidence to support this comes from the observation that the immunoglobulin or T-cell receptor antigen rearrangements that are unique to each patient's leukemia cells can be detected in blood samples obtained at birth.[
Evidence also exists that some children who never develop ALL are born with rare blood cells carrying a genomic alteration associated with ALL. Initial studies focused on the ETV6::RUNX1 translocation and used reverse transcriptase–polymerase chain reaction (PCR) to identify RNA transcripts indicating the presence of the gene fusion. For example, in one study, 1% of neonatal blood spots (Guthrie cards) tested positive for the ETV6::RUNX1 translocation.[
To more definitively address this question, a highly sensitive and specific DNA-based approach (genomic inverse PCR for exploration of ligated breakpoints) was applied to DNA from 1,000 cord blood specimens and found that 5% of specimens had the ETV6::RUNX1 translocation.[
Clinical Presentation
The typical and atypical symptoms and clinical findings of childhood ALL have been published.[
Diagnosis
The evaluation needed to definitively diagnose childhood ALL has been published.[
Overall Prognosis
Among children with ALL, approximately 98% attain remission. Approximately 85% of patients aged 1 to 18 years with newly diagnosed ALL treated on current regimens are expected to be long-term event-free survivors, with more than 90% of patients alive at 5 years.[
Cytogenetic and genomic findings combined with minimal residual disease (MRD) results can define subsets of ALL with EFS rates exceeding 95% and, conversely, subsets with EFS rates of 50% or lower. For more information, see the sections on Cytogenetics/Genomics of Childhood ALL and Prognostic Factors Affecting Risk-Based Treatment.
Despite the treatment advances in childhood ALL, numerous important biological and therapeutic questions remain to be answered before the goal of curing every child with ALL with the least associated toxicity can be achieved. The systematic investigation of these issues requires large clinical trials, and the opportunity to participate in these trials is offered to most patients and families.
Clinical trials for children and adolescents with ALL are generally designed to compare therapy that is currently accepted as standard with investigational regimens that seek to improve cure rates and/or decrease toxicity. In certain trials in which the cure rate for the patient group is very high, therapy reduction questions may be asked. Much of the progress made in identifying curative therapies for childhood ALL and other childhood cancers has been achieved through investigator-driven discovery and tested in carefully randomized, controlled, multi-institutional clinical trials. Information about ongoing clinical trials is available from the
Current Clinical Trials
Use our
References:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed March 6, 2024. - National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute.
Available online . Last accessed August 23, 2024. - Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28.
Also available online . Last accessed August 21, 2023. - Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29.
Also available online . Last accessed August 21, 2023. - Howlader N, Noone AM, Krapcho M: SEER Cancer Statistics Review (CSR) 1975-2013. Bethesda, Md: National Cancer Institute, 2015.
Available online . Last accessed June 04, 2021. - Surveillance, Epidemiology, and End Results Program: SEER Cancer Stat Facts: Childhood Leukemia (Ages 0–19). Bethesda, Md: National Cancer Institute, DCCPS, Surveillance Research Program.
Available online . Last accessed September 7, 2022. - Special section: cancer in children and adolescents. In: American Cancer Society: Cancer Facts and Figures 2014. American Cancer Society, 2014, pp 25-42.
Available online . Last accessed June 04, 2021. - Shah A, Coleman MP: Increasing incidence of childhood leukaemia: a controversy re-examined. Br J Cancer 97 (7): 1009-12, 2007.
- Smith MA, Ries LA, Gurney JG, et al.: Leukemia. In: Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649, pp 17-34.
Also available online . Last accessed August 11, 2022. - Barrington-Trimis JL, Cockburn M, Metayer C, et al.: Rising rates of acute lymphoblastic leukemia in Hispanic children: trends in incidence from 1992 to 2011. Blood 125 (19): 3033-4, 2015.
- Bennett JM, Catovsky D, Daniel MT, et al.: The morphological classification of acute lymphoblastic leukaemia: concordance among observers and clinical correlations. Br J Haematol 47 (4): 553-61, 1981.
- Koehler M, Behm FG, Shuster J, et al.: Transitional pre-B-cell acute lymphoblastic leukemia of childhood is associated with favorable prognostic clinical features and an excellent outcome: a Pediatric Oncology Group study. Leukemia 7 (12): 2064-8, 1993.
- Stiller CA, Chessells JM, Fitchett M: Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer 70 (5): 969-72, 1994.
- Passarge E: Bloom's syndrome: the German experience. Ann Genet 34 (3-4): 179-97, 1991.
- Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.
- Taylor AM, Metcalfe JA, Thick J, et al.: Leukemia and lymphoma in ataxia telangiectasia. Blood 87 (2): 423-38, 1996.
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
- Powell BC, Jiang L, Muzny DM, et al.: Identification of TP53 as an acute lymphocytic leukemia susceptibility gene through exome sequencing. Pediatr Blood Cancer 60 (6): E1-3, 2013.
- Hof J, Krentz S, van Schewick C, et al.: Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 29 (23): 3185-93, 2011.
- Ilencikova D, Sejnova D, Jindrova J, et al.: High-grade brain tumors in siblings with biallelic MSH6 mutations. Pediatr Blood Cancer 57 (6): 1067-70, 2011.
- Ripperger T, Schlegelberger B: Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet 59 (3): 133-42, 2016.
- Moriyama T, Relling MV, Yang JJ: Inherited genetic variation in childhood acute lymphoblastic leukemia. Blood 125 (26): 3988-95, 2015.
- Li Y, Schwab C, Ryan SL, et al.: Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508 (7494): 98-102, 2014.
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
- Hasle H: Pattern of malignant disorders in individuals with Down's syndrome. Lancet Oncol 2 (7): 429-36, 2001.
- Lupo PJ, Schraw JM, Desrosiers TA, et al.: Association Between Birth Defects and Cancer Risk Among Children and Adolescents in a Population-Based Assessment of 10 Million Live Births. JAMA Oncol 5 (8): 1150-1158, 2019.
- Marlow EC, Ducore J, Kwan ML, et al.: Leukemia Risk in a Cohort of 3.9 Million Children with and without Down Syndrome. J Pediatr 234: 172-180.e3, 2021.
- Brown AL, de Smith AJ, Gant VU, et al.: Inherited genetic susceptibility to acute lymphoblastic leukemia in Down syndrome. Blood 134 (15): 1227-1237, 2019.
- Zeller B, Gustafsson G, Forestier E, et al.: Acute leukaemia in children with Down syndrome: a population-based Nordic study. Br J Haematol 128 (6): 797-804, 2005.
- Arico M, Ziino O, Valsecchi MG, et al.: Acute lymphoblastic leukemia and Down syndrome: presenting features and treatment outcome in the experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Cancer 113 (3): 515-21, 2008.
- Maloney KW, Carroll WL, Carroll AJ, et al.: Down syndrome childhood acute lymphoblastic leukemia has a unique spectrum of sentinel cytogenetic lesions that influences treatment outcome: a report from the Children's Oncology Group. Blood 116 (7): 1045-50, 2010.
- de Graaf G, Buckley F, Skotko BG: Estimation of the number of people with Down syndrome in the United States. Genet Med 19 (4): 439-447, 2017.
- Chessells JM, Harrison G, Richards SM, et al.: Down's syndrome and acute lymphoblastic leukaemia: clinical features and response to treatment. Arch Dis Child 85 (4): 321-5, 2001.
- Buitenkamp TD, Izraeli S, Zimmermann M, et al.: Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood 123 (1): 70-7, 2014.
- Hertzberg L, Vendramini E, Ganmore I, et al.: Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the International BFM Study Group. Blood 115 (5): 1006-17, 2010.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Gaikwad A, Rye CL, Devidas M, et al.: Prevalence and clinical correlates of JAK2 mutations in Down syndrome acute lymphoblastic leukaemia. Br J Haematol 144 (6): 930-2, 2009.
- Kearney L, Gonzalez De Castro D, Yeung J, et al.: Specific JAK2 mutation (JAK2R683) and multiple gene deletions in Down syndrome acute lymphoblastic leukemia. Blood 113 (3): 646-8, 2009.
- Mullighan CG, Zhang J, Harvey RC, et al.: JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 106 (23): 9414-8, 2009.
- Hanada I, Terui K, Ikeda F, et al.: Gene alterations involving the CRLF2-JAK pathway and recurrent gene deletions in Down syndrome-associated acute lymphoblastic leukemia in Japan. Genes Chromosomes Cancer 53 (11): 902-10, 2014.
- Michels N, Boer JM, Enshaei A, et al.: Minimal residual disease, long-term outcome, and IKZF1 deletions in children and adolescents with Down syndrome and acute lymphocytic leukaemia: a matched cohort study. Lancet Haematol 8 (10): e700-e710, 2021.
- Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al.: Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1006-10, 2009.
- Treviño LR, Yang W, French D, et al.: Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet 41 (9): 1001-5, 2009.
- Migliorini G, Fiege B, Hosking FJ, et al.: Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood 122 (19): 3298-307, 2013.
- Hungate EA, Vora SR, Gamazon ER, et al.: A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun 7: 10635, 2016.
- Sherborne AL, Hosking FJ, Prasad RB, et al.: Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42 (6): 492-4, 2010.
- Xu H, Yang W, Perez-Andreu V, et al.: Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst 105 (10): 733-42, 2013.
- Ellinghaus E, Stanulla M, Richter G, et al.: Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia 26 (5): 902-9, 2012.
- Qian M, Zhao X, Devidas M, et al.: Genome-Wide Association Study of Susceptibility Loci for T-Cell Acute Lymphoblastic Leukemia in Children. J Natl Cancer Inst 111 (12): 1350-1357, 2019.
- Somasundaram R, Prasad MA, Ungerbäck J, et al.: Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126 (2): 144-52, 2015.
- Shah S, Schrader KA, Waanders E, et al.: A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 45 (10): 1226-31, 2013.
- Auer F, Rüschendorf F, Gombert M, et al.: Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia 28 (5): 1136-8, 2014.
- Zhang MY, Churpek JE, Keel SB, et al.: Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet 47 (2): 180-5, 2015.
- Topka S, Vijai J, Walsh MF, et al.: Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genet 11 (6): e1005262, 2015.
- Noetzli L, Lo RW, Lee-Sherick AB, et al.: Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet 47 (5): 535-8, 2015.
- Rampersaud E, Ziegler DS, Iacobucci I, et al.: Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv 3 (7): 1039-1046, 2019.
- Nishii R, Baskin-Doerfler R, Yang W, et al.: Molecular basis of ETV6-mediated predisposition to childhood acute lymphoblastic leukemia. Blood 137 (3): 364-373, 2021.
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.
- Churchman ML, Qian M, Te Kronnie G, et al.: Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 33 (5): 937-948.e8, 2018.
- Kuehn HS, Boisson B, Cunningham-Rundles C, et al.: Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med 374 (11): 1032-1043, 2016.
- Greaves MF, Wiemels J: Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer 3 (9): 639-49, 2003.
- Taub JW, Konrad MA, Ge Y, et al.: High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood 99 (8): 2992-6, 2002.
- Bateman CM, Colman SM, Chaplin T, et al.: Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 115 (17): 3553-8, 2010.
- Greaves MF, Maia AT, Wiemels JL, et al.: Leukemia in twins: lessons in natural history. Blood 102 (7): 2321-33, 2003.
- Mori H, Colman SM, Xiao Z, et al.: Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A 99 (12): 8242-7, 2002.
- Schäfer D, Olsen M, Lähnemann D, et al.: Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 131 (7): 821-826, 2018.
- Hein D, Dreisig K, Metzler M, et al.: The preleukemic TCF3-PBX1 gene fusion can be generated in utero and is present in ≈0.6% of healthy newborns. Blood 134 (16): 1355-1358, 2019.
- Gramatges MM, O'Brien MM, Rabin KR: Acute lymphoblastic leukemia. In: Blaney SM, Helman LJ, Adamson PC, eds.: Pizzo and Poplack's Pediatric Oncology. 8th ed. Wolters Kluwer, 2020, pp 419-53.
- Chessells JM; haemostasis and thrombosis task force, British committee for standards in haematology: Pitfalls in the diagnosis of childhood leukaemia. Br J Haematol 114 (3): 506-11, 2001.
- Onciu M: Acute lymphoblastic leukemia. Hematol Oncol Clin North Am 23 (4): 655-74, 2009.
- Margolskee E, Waith Wertheim GB, Harvey RC: Pathology and molecular diagnosis of leukemias and lymphomas. In: Blaney SM, Helman LJ, Adamson PC, eds.: Pizzo and Poplack's Pediatric Oncology. 8th ed. Wolters Kluwer, 2020, pp 117-30.
- Cheng J, Klairmont MM, Choi JK: Peripheral blood flow cytometry for the diagnosis of pediatric acute leukemia: Highly reliable with rare exceptions. Pediatr Blood Cancer 66 (1): e27453, 2019.
- Möricke A, Zimmermann M, Valsecchi MG, et al.: Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood 127 (17): 2101-12, 2016.
- Vora A, Goulden N, Wade R, et al.: Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol 14 (3): 199-209, 2013.
- Place AE, Stevenson KE, Vrooman LM, et al.: Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, open-label phase 3 trial. Lancet Oncol 16 (16): 1677-90, 2015.
- Pieters R, de Groot-Kruseman H, Van der Velden V, et al.: Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol 34 (22): 2591-601, 2016.
- Moorman AV, Antony G, Wade R, et al.: Time to Cure for Childhood and Young Adult Acute Lymphoblastic Leukemia Is Independent of Early Risk Factors: Long-Term Follow-Up of the UKALL2003 Trial. J Clin Oncol 40 (36): 4228-4239, 2022.
Cytogenetics / Genomics of Childhood ALL
Genomics of childhood ALL
The genomics of childhood acute lymphoblastic leukemia (ALL) has been extensively investigated, and multiple distinctive subtypes have been defined on the basis of cytogenetic and molecular characterizations, each with its own pattern of clinical and prognostic characteristics.[
Throughout this section, the percentages of genomic subtypes from among all B-ALL and T-ALL cases are derived primarily from a report describing the genomic characterization of patients treated on several Children's Oncology Group (COG) and St. Jude Children's Research Hospital (SJCRH) clinical trials. Percentages by subtype are presented for NCI standard-risk and NCI high-risk patients with B-ALL (up to age 18 years).[
B-ALL cytogenetics/genomics
B-ALL is typified by genomic alterations that include: 1) gene fusions that lead to aberrant activity of transcription factors, 2) chromosomal gains and losses (e.g., hyperdiploidy or hypodiploidy), and 3) alterations leading to activation of tyrosine kinase genes.[
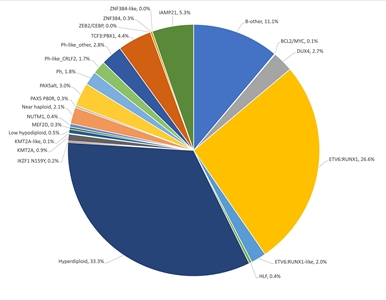
Figure 2. Genomic subtypes and frequencies of NCI standard-risk B-ALL. The figure represents data from 1,126 children diagnosed with NCI standard-risk B-ALL (aged 1–9 years and WBC <50,000/µL) and enrolled in St. Jude Children's Research Hospital or Children's Oncology Group clinical trials. Adapted from Supplemental Table 2 of Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
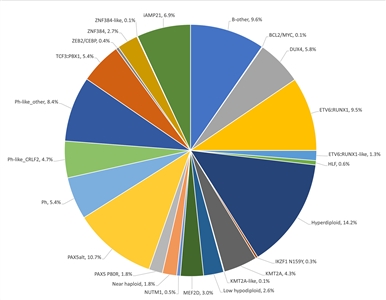
Figure 3. Genomic subtypes and frequencies of NCI high-risk B-ALL. The figure represents data from 1,084 children diagnosed with NCI high-risk B-ALL (aged 1–18 years and WBC >50,000/µL) and enrolled in St. Jude Children's Research Hospital or Children's Oncology Group clinical trials. Adapted from Supplemental Table 2 of Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
The genomic landscape of B-ALL is characterized by a range of genomic alterations that disrupt normal B-cell development and, in some cases, by variants in genes that provide a proliferation signal (e.g., activating variants in RAS family genes or variants/translocations leading to kinase pathway signaling). Genomic alterations leading to blockage of B-cell development include translocations (e.g., TCF3::PBX1 and ETV6::RUNX1 fusions), point variants (e.g., IKZF1 and PAX5), and intragenic/intergenic deletions (e.g., IKZF1, PAX5, EBF, and ERG).[
The genomic alterations in B-ALL tend not to occur at random, but rather to cluster within subtypes that can be delineated by biological characteristics such as their gene expression profiles. Cases with recurring chromosomal translocations (e.g., TCF3::PBX1 and ETV6::RUNX1 fusions and KMT2A-rearranged ALL) have distinctive biological features and illustrate this point, as do the examples below of specific genomic alterations within unique biological subtypes:
- IKZF1 deletions and variants are most commonly observed within cases of BCR::ABL1 ALL and BCR::ABL1-like ALL.[
3 ,4 ] - Intragenic ERG deletions occur within a distinctive subtype characterized by gene rearrangements involving DUX4.[
5 ,6 ] - TP53 variants, often germline, occur at high frequency in patients with low hypodiploid ALL with 32 to 39 chromosomes.[
7 ]TP53 variants are uncommon in other patients with B-ALL.
Activating point variants in kinase genes are uncommon in high-risk B-ALL. JAK genes are the primary kinases that are found to be altered. These variants are generally observed in patients with BCR::ABL1-like ALL who have CRLF2 abnormalities, although JAK2 variants are also observed in approximately 25% of children with Down syndrome and ALL, occurring exclusively in cases with CRLF2 gene rearrangements.[
Understanding of the genomics of B-ALL at relapse is less advanced than the understanding of ALL genomics at diagnosis. Childhood ALL is often polyclonal at diagnosis and under the selective influence of therapy, some clones may be extinguished and new clones with distinctive genomic profiles may arise.[
Several recurrent chromosomal abnormalities have been shown to have prognostic significance, especially in B-ALL. Some chromosomal alterations are associated with more favorable outcomes, such as favorable trisomies (51–65 chromosomes) and the ETV6::RUNX1 fusion.[
In recognition of the clinical significance of many of these genomic alterations, the 5th edition revision of the World Health Organization Classification of Haematolymphoid Tumours lists the following entities for B-ALL:[
- B-lymphoblastic leukemia/lymphoma, NOS.
- B-lymphoblastic leukemia/lymphoma with high hyperdiploidy.
- B-lymphoblastic leukemia/lymphoma with hypodiploidy.
- B-lymphoblastic leukemia/lymphoma with iAMP21.
- B-lymphoblastic leukemia/lymphoma with BCR::ABL1 fusion.
- B-lymphoblastic leukemia/lymphoma with BCR::ABL1-like features.
- B-lymphoblastic leukemia/lymphoma with KMT2A rearrangement.
- B-lymphoblastic leukemia/lymphoma with ETV6::RUNX1 fusion.
- B-lymphoblastic leukemia/lymphoma with ETV6::RUNX1-like features.
- B-lymphoblastic leukemia/lymphoma with TCF3::PBX1 fusion.
- B-lymphoblastic leukemia/lymphoma with IGH::IL3 fusion.
- B-lymphoblastic leukemia/lymphoma with TCF3::HLF fusion.
- B-lymphoblastic leukemia/lymphoma with other defined genetic abnormalities.
The category of B-ALL with other defined genetic abnormalities includes potential novel entities, including B-ALL with DUX4, MEF2D, ZNF384 or NUTM1 rearrangements; B-ALL with IG::MYC fusions; and B-ALL with PAX5alt or PAX5 p.P80R (NP_057953.1) abnormalities.
These and other chromosomal and genomic abnormalities for childhood ALL are described below.
- Chromosome number.
- High hyperdiploidy (51–65 chromosomes).
High hyperdiploidy, defined as 51 to 65 chromosomes per cell or a DNA index greater than 1.16, occurs in approximately 33% of NCI standard-risk and 14% of NCI high-risk pediatric B-ALL cases.[
1 ,19 ] Hyperdiploidy can be evaluated by measuring the DNA content of cells (DNA index) or by karyotyping. In cases with a normal karyotype or in which standard cytogenetic analysis was unsuccessful, interphase fluorescence in situ hybridization (FISH) may detect hidden hyperdiploidy.High hyperdiploidy generally occurs in cases with clinically favorable prognostic factors (patients aged 1 to <10 years with a low white blood cell [WBC] count) and is an independent favorable prognostic factor.[
19 ,20 ,21 ] Within the hyperdiploid range of 51 to 65 chromosomes, patients with higher modal numbers (58–66) appeared to have a better prognosis in one study.[21 ] Hyperdiploid leukemia cells are particularly susceptible to undergoing apoptosis and accumulate higher levels of methotrexate and its active polyglutamate metabolites,[22 ] which may explain the favorable outcome commonly observed in these cases.While the overall outcome of patients with high hyperdiploidy is considered to be favorable, factors such as age, WBC count, specific trisomies, and early response to treatment have been shown to modify its prognostic significance.[
23 ,24 ]Multiple reports have described the prognostic significance of specific chromosome trisomies among children with hyperdiploid B-ALL.
- A study combining experience from the Children's Cancer Group and the Pediatric Oncology Group (POG) found that patients with trisomies of chromosomes 4, 10, and 17 (triple trisomies) have a particularly favorable outcome.[
25 ]; [16 ][Level of evidence B4] - A report using POG data found that NCI standard-risk patients with trisomies of 4 and 10, without regard to chromosome 17 status, have an excellent prognosis.[
26 ] COG protocols currently use double trisomies of chromosomes 4 and 10 to define favorable hyperdiploidy. - A retrospective analysis evaluated patients treated on two consecutive UKALL trials to identify and validate a profile to predict outcome in high hyperdiploid B-ALL. The investigators defined a good-risk group (approximately 80% of high hyperdiploidy patients) that was associated with a more favorable prognosis. Good-risk patients had either trisomies of both chromosomes 17 and 18 or trisomy of one of these two chromosomes along with absence of trisomies of chromosomes 5 and 20. All other patients were defined as poor risk and had a less favorable outcome. End-induction MRD and copy number alterations (such as IKZF1 deletion) were prognostically significant within each hyperdiploid risk group.[
27 ]
Chromosomal translocations may be seen with high hyperdiploidy, and in those cases, patients are more appropriately risk-classified on the basis of the prognostic significance of the translocation. For instance, in one study, 8% of patients with the BCR::ABL1 fusion also had high hyperdiploidy,[
28 ] and the outcome of these patients (treated without tyrosine kinase inhibitors) was inferior to that observed in non-BCR::ABL1 high hyperdiploid patients.Certain patients with hyperdiploid ALL may have a hypodiploid clone that has doubled (masked hypodiploidy).[
29 ] Molecular technologies, such as single nucleotide polymorphism microarrays to detect widespread loss of heterozygosity, can be used to identify patients with masked hypodiploidy.[29 ] These cases may be interpretable based on the pattern of gains and losses of specific chromosomes (hyperdiploidy with two and four copies of chromosomes rather than three copies). These patients have an unfavorable outcome, similar to those with hypodiploidy.[30 ]Near triploidy (68–80 chromosomes) and near tetraploidy (>80 chromosomes) are much less common and appear to be biologically distinct from high hyperdiploidy.[
31 ] Unlike high hyperdiploidy, a high proportion of near tetraploid cases harbor a cryptic ETV6::RUNX1 fusion.[31 ,32 ,33 ] Near triploidy and tetraploidy were previously thought to be associated with an unfavorable prognosis, but later studies suggest that this may not be the case.[31 ,33 ]The genomic landscape of hyperdiploid ALL is characterized by variants in genes of the receptor tyrosine kinase (RTK)/RAS pathway in approximately one-half of cases. Genes encoding histone modifiers are also present in a recurring manner in a minority of cases. Analysis of variant profiles demonstrates that chromosomal gains are early events in the pathogenesis of hyperdiploid ALL and may occur in utero, while variants in RTK/RAS pathway genes are late events in leukemogenesis and are often subclonal.[
1 ,34 ] - A study combining experience from the Children's Cancer Group and the Pediatric Oncology Group (POG) found that patients with trisomies of chromosomes 4, 10, and 17 (triple trisomies) have a particularly favorable outcome.[
- Hypodiploidy (<44 chromosomes).
B-ALL cases with fewer than the normal number of chromosomes have been subdivided in various ways, with one report stratifying on the basis of modal chromosome number into the following four groups:[
30 ]- Near-haploid: 24 to 29 chromosomes (n = 46).
- Low-hypodiploid: 33 to 39 chromosomes (n = 26).
- High-hypodiploid: 40 to 43 chromosomes (n = 13).
- Near-diploid: 44 chromosomes (n = 54).
Near-haploid cases represent approximately 2% of NCI standard-risk and 2% of NCI high-risk pediatric B-ALL.[
1 ]Low-hypodiploid cases represent approximately 0.5% of NCI standard-risk and 2.6% of NCI high-risk pediatric B-ALL cases.[
1 ]Most patients with hypodiploidy are in the near-haploid and low-hypodiploid groups, and both of these groups have an elevated risk of treatment failure compared with nonhypodiploid cases.[
30 ,35 ] Patients with fewer than 44 chromosomes have a worse outcome than do patients with 44 or 45 chromosomes in their leukemic cells.[30 ] Several studies have shown that patients with high minimal residual disease (MRD) (≥0.01%) after induction do very poorly, with 5-year event-free survival (EFS) rates ranging from 25% to 47%. Although hypodiploid patients with low MRD after induction fare better (5-year EFS rates, 64%–75%), their outcomes are still inferior to most children with other types of ALL.[36 ,37 ,38 ]The recurring genomic alterations of near-haploid and low-hypodiploid ALL appear to be distinctive from each other and from other types of ALL.[
7 ] In near-haploid ALL, alterations targeting RTK signaling, RAS signaling, and IKZF3 are common.[39 ] In low-hypodiploid ALL, genetic alterations involving TP53, RB1, and IKZF2 are common. Importantly, the TP53 alterations observed in low-hypodiploid ALL are also present in nontumor cells in approximately 40% of cases, suggesting that these variants are germline and that low-hypodiploid ALL represents, in some cases, a manifestation of Li-Fraumeni syndrome.[7 ] Approximately two-thirds of patients with ALL and germline pathogenic TP53 variants have hypodiploid ALL.[40 ]
- High hyperdiploidy (51–65 chromosomes).
- Chromosomal translocations and gains/deletions of chromosomal segments.
- ETV6::RUNX1 fusion (t(12;21)(p13.2;q22.1)).
Fusion of the ETV6 gene on chromosome 12 to the RUNX1 gene on chromosome 21 is present in approximately 27% of NCI standard-risk and 10% of NCI high-risk pediatric B-ALL cases.[
1 ,32 ]The ETV6::RUNX1 fusion produces a cryptic translocation that is detected by methods such as FISH, rather than conventional cytogenetics, and it occurs most commonly in children aged 2 to 9 years.[
41 ,42 ] Hispanic children with ALL have a lower incidence of ETV6::RUNX1 fusions than do White children.[43 ]Reports generally indicate favorable EFS and overall survival (OS) rates in children with the ETV6::RUNX1 fusion; however, the prognostic impact of this genetic feature is modified by the following factors:[
44 ,45 ,46 ,47 ,48 ]; [16 ][Level of evidence B4]- Early response to treatment.
- NCI risk category (age and WBC count at diagnosis).
- Treatment regimen.
In one study of the treatment of newly diagnosed children with ALL, multivariate analysis of prognostic factors found age and leukocyte count, but not ETV6::RUNX1 fusion status, to be independent prognostic factors.[
44 ] However, another large trial only enrolled patients classified as having favorable-risk B-ALL, with low-risk clinical features, either trisomies of 4, 10, and 17 or ETV6::RUNX1 fusion, and end induction MRD less than 0.01%. Patients had a 5-year continuous complete remission rate of 93.7% and a 6-year OS rate of 98.2% for patients with ETV6::RUNX1.[16 ] It does not appear that the presence of secondary cytogenetic abnormalities, such as deletion of ETV6 (12p) or CDKN2A/B (9p), impacts the outcome of patients with the ETV6::RUNX1 fusion.[48 ,49 ]There is a higher frequency of late relapses in patients with ETV6::RUNX1 fusions compared with other relapsed B-ALL patients.[
44 ,50 ] Patients with the ETV6::RUNX1 fusion who relapse seem to have a better outcome than other relapse patients,[51 ] with an especially favorable prognosis for patients who relapse more than 36 months from diagnosis.[52 ] Some relapses in patients with ETV6::RUNX1 fusions may represent a new independent second hit in a persistent preleukemic clone (with the first hit being the ETV6::RUNX1 translocation).[53 ,54 ] - BCR::ABL1 fusion (t(9;22)(q34.1;q11.2); Ph+).
The BCR::ABL1 fusion leads to production of a BCR::ABL1 fusion protein with tyrosine kinase activity (see Figure 4).[
1 ] The BCR::ABL1 fusion occurs in approximately 2% of NCI standard-risk and 5% of NCI high-risk pediatric B-ALL cases.[1 ] The BCR::ABL1 fusion is also the leukemogenic driver for chronic myeloid leukemia (CML). The most common BCR breakpoint in CML is different from the most common BCR breakpoint in ALL. The breakpoint that typifies CML produces a larger fusion protein (termed p210) than the breakpoint most commonly observed for ALL (termed p190, a smaller fusion protein).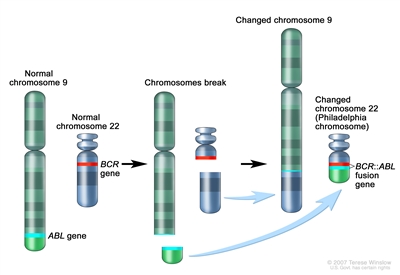
Figure 4. The Philadelphia chromosome is a translocation between the ABL1 oncogene (on the long arm of chromosome 9) and the BCR gene (on the long arm of chromosome 22), resulting in the fusion gene BCR::ABL1. BCR::ABL1 encodes an oncogenic protein with tyrosine kinase activity.Ph+ ALL is more common in older children with B-ALL and high WBC counts, with the incidence of the BCR::ABL1 fusions increasing to about 25% in young adults with ALL.
Historically, the BCR::ABL1 fusion was associated with an extremely poor prognosis (especially in those who presented with a high WBC count or had a slow early response to initial therapy), and its presence had been considered an indication for allogeneic hematopoietic stem cell transplant (HSCT) in patients in first remission.[
28 ,55 ,56 ,57 ] Inhibitors of the BCR::ABL1 tyrosine kinase, such as imatinib mesylate, are effective in patients with BCR::ABL1 ALL.[58 ] A study by the Children's Oncology Group (COG), which used intensive chemotherapy and concurrent imatinib mesylate given daily, demonstrated a 5-year EFS rate of 70% (± 12%), which was superior to the EFS rate of historical controls in the pre-tyrosine kinase inhibitor (imatinib mesylate) era. This result eliminated the recommendation of HSCT for patients with a good early response to chemotherapy using a tyrosine kinase inhibitor.[59 ,60 ]The International Consensus Classification of acute lymphoblastic leukemia/lymphoma from 2022 divides BCR::ABL1–positive B-ALL into two subtypes: cases with lymphoid-only involvement and cases with multilineage involvement.[
61 ] These subtypes differ in the timing of their transformation event. A multipotent progenitor serves as the target cell of origin for BCR::ABL1–positive B-ALL with multilineage involvement, and a later progenitor is the target cell of origin for BCR::ABL1–positive B-ALL with lymphoid-only involvement.- BCR::ABL1–positive B-ALL with lymphoid-only involvement is the predominate subtype. Only a minority of cases in children and adults have multilineage involvement (estimated at 15%–30%).[
62 ] - BCR::ABL1–positive B-ALL cases with lymphoid-only involvement and cases with multilineage involvement have similar clinical presentations and immunophenotypes. In addition, both subtypes commonly have the p190 fusion protein.[
62 ,63 ] - One way of distinguishing between patients with lymphoid-only and multilineage involvement is to detect the BCR::ABL1 fusion in normal non-ALL B cells, T cells, and myeloid cells.[
63 ] - A second way of distinguishing between patients with lymphoid-only and multilineage involvement is to detect quantitative differences in MRD levels (typically 1 log) using measures that quantify BCR::ABL1 DNA or RNA, compared with measures based on flow cytometry, real-time quantitative polymerase chain reaction (PCR), or next-generation sequencing (NGS) quantitation of leukemia-specific immunoglobulin (IG) or T-cell receptor (TCR) rearrangements.[
62 ,63 ,64 ]- For patients with lymphoid-only BCR::ABL1–positive B-ALL, MRD estimates for these methods will be correlated with each other.
- For patients with multilineage involvement BCR::ABL1–positive B-ALL, posttreatment MRD estimates based on detection of BCR::ABL1 DNA or RNA will often be higher than estimates based on flow cytometry or quantitation of leukemia-specific IG/TCR rearrangements.
- For patients with BCR::ABL1–positive B-ALL and multilineage involvement, levels of BCR::ABL1 transcripts and DNA may remain stable over time despite continued treatment with chemotherapy and tyrosine kinase inhibitors. In these situations, the persisting BCR::ABL1 DNA or RNA likely represents evidence of a residual preleukemic clone and not leukemia cells. Therefore, the term MRD is a misnomer.
- A corollary of the difference in MRD detection by methods based on BCR::ABL1 DNA or RNA detection versus MRD detection based on flow cytometry or IG/TCR rearrangements is that the latter methods provide more reliable prognostication.[
62 ,64 ,65 ] For example, the presence of MRD by BCR::ABL1 DNA or RNA detection in the absence of MRD detection by IG/TCR rearrangements does not confer inferior prognosis. - Based on the limited numbers of patients studied to date, prognosis appears similar in both adults and children with lymphoid-only versus multilineage involvement BCR::ABL1–positive B-ALL.[
62 ,64 ] - There are case reports of patients with multilineage involvement BCR::ABL1–positive B-ALL who relapse years from their initial diagnosis. In addition, their relapsed leukemia has the same BCR::ABL1 breakpoint as their initial leukemia, but it has a different IG/TCR rearrangement.[
64 ] These case reports suggest that patients with multilineage BCR::ABL1–positive B-ALL are at risk of a second leukemogenic event, leading to a second BCR::ABL1 leukemia. - There is no evidence that a specific monitoring schedule or prolonged treatment with a tyrosine kinase inhibitor provides clinical benefit for patients with multilineage involvement BCR::ABL1–positive B-ALL who have maintained presence of BCR::ABL1 transcripts or DNA at the completion of a standard-duration course of leukemia therapy.
- BCR::ABL1–positive B-ALL with lymphoid-only involvement is the predominate subtype. Only a minority of cases in children and adults have multilineage involvement (estimated at 15%–30%).[
- KMT2A-rearranged ALL (t(v;11q23.3)).
Rearrangements involving the KMT2A gene with more than 100 translocation partner genes result in the production of fusion oncoproteins. KMT2A gene rearrangements occur in up to 80% of infants with ALL. Beyond infancy, approximately 1% of NCI standard-risk and 4% of NCI high-risk pediatric B-ALL cases have KMT2A rearrangements.[
1 ]These rearrangements are generally associated with an increased risk of treatment failure, particularly in infants.[
66 ,67 ,68 ,69 ] The KMT2A::AFF1 fusion (t(4;11)(q21;q23)) is the most common rearrangement involving the KMT2A gene in children with ALL and occurs in approximately 1% to 2% of childhood ALL.[67 ,70 ]Patients with KMT2A::AFF1 fusions are usually infants with high WBC counts. These patients are more likely than other children with ALL to have central nervous system (CNS) disease and to have a poor response to initial therapy.[
71 ] While both infants and adults with the KMT2A::AFF1 fusion are at high risk of treatment failure, children with the KMT2A::AFF1 fusion appear to have a better outcome.[66 ,67 ,72 ] Irrespective of the type of KMT2A gene rearrangement, infants with KMT2A-rearranged ALL have much worse event-free survival rates than non-infant pediatric patients with KMT2A-rearranged ALL.[66 ,67 ,72 ]Whole-genome sequencing has determined that cases of infant ALL with KMT2A gene rearrangements have frequent subclonal NRAS or KRAS variants and few additional genomic alterations, none of which have clear clinical significance.[
11 ,73 ] Deletion of the KMT2A gene has not been associated with an adverse prognosis.[74 ]Of interest, the KMT2A::MLLT1 fusion (t(11;19)(q23;p13.3)) occurs in approximately 1% of ALL cases and occurs in both early B-lineage ALL and T-ALL.[
75 ] Outcome for infants with the KMT2A::MLLT1 fusion is poor, but outcome appears relatively favorable in older children with T-ALL and the KMT2A::MLLT1 fusion.[75 ] - TCF3::PBX1 fusion (t(1;19)(q23;p13.3)) and TCF3::HLF fusion (t(17;19)(q22;p13)).
Fusion of the TCF3 gene on chromosome 19 to the PBX1 gene on chromosome 1 is present in approximately 4% of NCI standard-risk and 5% of NCI high-risk pediatric B-ALL cases.[
1 ,76 ,77 ] The TCF3::PBX1 fusion may occur as either a balanced translocation or as an unbalanced translocation and is the primary recurring genomic alteration of the pre-B–ALL immunophenotype (cytoplasmic immunoglobulin positive).[78 ] Black children are relatively more likely than White children to have pre-B–ALL with the TCF3::PBX1 fusion.[79 ]The TCF3::PBX1 fusion had been associated with inferior outcome in the context of antimetabolite-based therapy,[
80 ] but the adverse prognostic significance was largely negated by more aggressive multiagent therapies.[77 ,81 ] More specifically, in a trial conducted by St. Jude Children's Research Hospital (SJCRH) in which all patients were treated without cranial radiation, patients with the TCF3::PBX1 fusion had an overall outcome comparable to children lacking this translocation, but with a higher risk of CNS relapse and a lower rate of bone marrow relapse, suggesting that more intensive CNS therapy may be needed for these patients.[82 ,83 ]The TCF3::HLF fusion occurs in less than 1% of pediatric ALL cases. ALL with the TCF3::HLF fusion is associated with disseminated intravascular coagulation and hypercalcemia at diagnosis. Outcome is very poor for children with the TCF3::HLF fusion, with a literature review noting mortality for 20 of 21 cases reported.[
84 ] In addition to the TCF3::HLF fusion, the genomic landscape of this ALL subtype was characterized by deletions in genes involved in B-cell development (PAX5, BTG1, and VPREB1) and by variants in RAS pathway genes (NRAS, KRAS, and PTPN11).[78 ] - DUX4-rearranged ALL with frequent ERG deletions.
Approximately 3% of NCI standard-risk and 6% of NCI high-risk pediatric B-ALL patients have a rearrangement involving DUX4 that leads to its overexpression.[
1 ,5 ,6 ] East Asian ancestry was linked to an increased prevalence of DUX4-rearranged ALL (favorable).[85 ] The most common rearrangement produces IGH::DUX4 fusions, with ERG::DUX4 fusions also observed.[86 ]DUX4-rearranged cases show a distinctive gene expression pattern that was initially identified as being associated with focal deletions in ERG,[86 ,87 ,88 ,89 ] and one-half to more than two-thirds of these cases have focal intragenic deletions involving ERG that are not observed in other ALL subtypes.[5 ,86 ]ERG deletions often appear to be clonal, but using sensitive detection methodology, it appears that most cases are polyclonal.[86 ]IKZF1 alterations are observed in 20% to 40% of DUX4-rearranged ALL.[5 ,6 ]ERG deletion connotes an excellent prognosis, with OS rates exceeding 90%. Even when the IZKF1 deletion is present, prognosis remains highly favorable.[
87 ,88 ,89 ,90 ] While patients with DUX4-rearranged ALL have an overall favorable prognosis, there is uncertainty as to whether this applies to both ERG-deleted and ERG-intact cases. In a study of 50 patients with DUX4-rearranged ALL, patients with an ERG deletion detected by genomic PCR (n = 33) had a more favorable EFS rate of approximately 90% than did patients with intact ERG (n = 17), with an EFS rate of approximately 70%.[88 ] - MEF2D-rearranged ALL.
Gene fusions involving MEF2D, a transcription factor that is expressed during B-cell development, are observed in approximately 0.3% of NCI standard-risk and 3% of NCI high-risk pediatric B-ALL cases.[
1 ,91 ,92 ]Although multiple fusion partners may occur, most cases involve BCL9, which is located on chromosome 1q21, as is MEF2D.[
91 ,93 ] The interstitial deletion producing the MEF2D::BCL9 fusion is too small to be detected by conventional cytogenetic methods. Cases with MEF2D gene fusions show a distinctive gene expression profile, except for rare cases with MEF2D::CSFR1 that have a BCR::ABL1-like gene expression profile.[91 ,94 ]The median age at diagnosis for cases of MEF2D-rearranged ALL in studies that included both adult and pediatric patients was 12 to 14 years.[
91 ,92 ] For 22 children with MEF2D-rearranged ALL enrolled in a high-risk ALL clinical trial, the 5-year EFS rate was 72% (standard error, ± 10%), which was inferior to that for other patients.[91 ] - ZNF384-rearranged ALL.
ZNF384 is a transcription factor that is rearranged in approximately 0.3% of NCI standard-risk and 2.7% of NCI high-risk pediatric B-ALL cases.[
1 ,91 ,95 ,96 ]East Asian ancestry was associated with an increased prevalence of ZNF384.[
85 ] Multiple fusion partners for ZNF384 have been reported, including ARID1B, CREBBP, EP300, SMARCA2, TAF15, and TCF3. Regardless of the fusion partner, ZNF384-rearranged ALL cases show a distinctive gene expression profile.[91 ,95 ,96 ]ZNF384 rearrangement does not appear to confer independent prognostic significance.[91 ,95 ,96 ] However, within the subset of patients with ZNF384 rearrangements, patients with EP300::ZNF384 fusions have lower relapse rates than patients with other ZNF384 fusion partners.[97 ] The immunophenotype of B-ALL with ZNF384 rearrangement is characterized by weak or negative CD10 expression, with expression of CD13 and/or CD33 commonly observed.[95 ,96 ] Cases of mixed phenotype acute leukemia (MPAL) (B/myeloid) that have ZNF384 gene fusions have been reported,[98 ,99 ] and a genomic evaluation of MPAL found that ZNF384 gene fusions were present in approximately one-half of B/myeloid cases.[100 ] - NUTM1-rearranged B-ALL.
NUTM1-rearranged B-ALL is most commonly observed in infants, representing 3% to 5% of overall cases of B-ALL in this age group and approximately 20% of infant B-ALL cases lacking the KMT2A rearrangement.[
101 ] The frequency of NUTM1 rearrangement is lower in children after infancy (<1% of cases).[1 ,101 ]The NUTM1 gene is located on chromosome 15q14, and some cases of B-ALL with NUTM1 rearrangements show chromosome 15q aberrations, but other cases are cryptic and have no cytogenetic abnormalities.[
102 ] RNA sequencing, as well as break-apart FISH, can be used to detect the presence of the NUTM1 rearrangement.[101 ]The NUTM1 rearrangement appears to be associated with a favorable outcome.[
101 ,103 ] Among 35 infants with NUTM1-rearranged B-ALL who were treated on Interfant protocols, all patients achieved remission and no relapses were observed.[101 ] For the 32 children older than 12 months with NUTM1-rearranged B-ALL, the 4-year EFS and OS rates were 92% and 100%, respectively. - IGH::IL3 fusion (t(5;14)(q31.1;q32.3)).
This entity is included in the 2016 revision of the World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues.[
104 ] The finding of t(5;14)(q31.1;q32.3) in patients with ALL and hypereosinophilia in the 1980s was followed by the identification of the IGH::IL3 fusion as the underlying genetic basis for the condition.[105 ,106 ] The joining of the IGH locus to the promoter region of the IL3 gene leads to dysregulation of IL3 expression.[107 ] Cytogenetic abnormalities in children with ALL and eosinophilia are variable, with only a subset resulting from the IGH::IL3 fusion.[108 ]The number of cases of IGH::IL3 ALL described in the published literature is too small to assess the prognostic significance of the IGH::IL3 fusion. Diagnosis of cases of IGH::IL3 ALL may be delayed because the ALL clone in the bone marrow may be small, and because it can present with hypereosinophilia in the absence of cytopenias and circulating blasts.[
104 ] - Intrachromosomal amplification of chromosome 21 (iAMP21).
iAMP21 occurs in approximately 5% of NCI standard-risk and 7% of NCI high-risk pediatric B-ALL cases.[
1 ] iAMP21 is generally diagnosed using FISH and is defined by the presence of greater than or equal to five RUNX1 signals per cell (or ≥3 extra copies of RUNX1 on a single abnormal chromosome).[104 ] iAMP21 can also be identified by chromosomal microarray analysis. Uncommonly, iAMP21 with an atypical genomic pattern (e.g., amplification of the genomic region but with less than 5 RUNX1 signals or having at least 5 RUNX1 signals with some located apart from the abnormal iAMP21-chromosome) is identified by microarray but not RUNX1 FISH.[109 ] The prognostic significance of iAMP21 defined only by microarray has not been characterized.iAMP21 is associated with older age (median, approximately 10 years), presenting WBC count of less than 50 × 109 /L, a slight female preponderance, and high end-induction MRD.[
110 ,111 ,112 ] Analysis of variant signatures indicates that gene amplifications in iAMP21 occur later in leukemogenesis, which is in contrast to those of hyperdiploid ALL that can arise early in life and even in utero.[1 ]The United Kingdom Acute Lymphoblastic Leukaemia (UKALL) clinical trials group initially reported that the presence of iAMP21 conferred a poor prognosis in patients treated in the MRC ALL 97/99 trial (5-year EFS rate, 29%).[
17 ] In their subsequent trial (UKALL2003 [NCT00222612]), patients with iAMP21 were assigned to a more intensive chemotherapy regimen and had a markedly better outcome (5-year EFS rate, 78%).[111 ] Similarly, the COG has reported that iAMP21 was associated with a significantly inferior outcome in NCI standard-risk patients (4-year EFS rate, 73% for iAMP21 vs. 92% in others), but not in NCI high-risk patients (4-year EFS rate, 73% vs. 80%).[110 ] On multivariate analysis, iAMP21 was an independent predictor of inferior outcome only in NCI standard-risk patients.[110 ] The results of the UKALL2003 and COG studies suggest that treatment of iAMP21 patients with high-risk chemotherapy regimens abrogates its adverse prognostic significance and obviates the need for HSCT in first remission.[112 ] - PAX5 alterations.
Gene expression analysis identified two distinctive ALL subsets with PAX5 genomic alterations, called PAX5alt and PAX5 p.P80R (NP_057953.1).[
113 ] The alterations in the PAX5alt subtype included rearrangements, sequence variants, and focal intragenic amplifications.PAX5alt. PAX5 rearrangements have been reported to represent approximately 3% of NCI standard-risk and 11% of NCI high-risk pediatric B-ALL cases.[
1 ] More than 20 partner genes for PAX5 have been described,[113 ] with PAX5::ETV6, the primary genomic alteration in dic(9;12)(p13;p13),[114 ] being the most common gene fusion.[113 ]Intragenic amplification of PAX5 was identified in approximately 1% of B-ALL cases, and it was usually detected in cases lacking known leukemia-driver genomic alterations.[
115 ] Cases with PAX5 amplification show male predominance (66%), with most (55%) having NCI high-risk status. For a cohort of patients with PAX5 amplification diagnosed between 1993 and 2015, the 5-year EFS rate was 49% (95% confidence interval [CI], 36%–61%), and the OS rate was 67% (95% CI, 54%–77%), suggesting a relatively poor prognosis for patients with this B-ALL subtype.PAX5 p.P80R (NP_057953.1). PAX5 with a p.P80R variant shows a gene expression profile distinctive from that of other cases with PAX5 alterations.[
113 ] Cases with PAX5 p.P80R represent approximately 0.3% of NCI standard-risk and 1.8% of NCI high-risk pediatric B-ALL.[1 ] PAX5 p.P80R B-ALL appears to occur more frequently in the adolescent and young adult (AYA) and adult populations (3.1% and 4.2%, respectively).[113 ]Outcome for the pediatric patients with PAX5 p.P80R and PAX5alt treated in a COG clinical trial appears to be intermediate (5-year EFS rate, approximately 75%).[
113 ]PAX5alt rearrangements have also been detected in infant patients with ALL, with a reported outcome similar to KMT2A-rearranged infant ALL.[103 ] - BCR::ABL1-like (Ph-like).
BCR::ABL1-negative patients with a gene expression profile similar to BCR::ABL1-positive patients have been referred to as Ph-like,[
116 ,117 ,118 ] and are now referred to as BCR::ABL1-like.[18 ] This occurs in 10% to 20% of pediatric B-ALL patients, increasing in frequency with age, and has been associated with an IKZF1 deletion or variant.[1 ,8 ,116 ,117 ,119 ,120 ]Retrospective analyses have indicated that patients with BCR::ABL1-like ALL have a poor prognosis.[
4 ,116 ] In one series, the 5-year EFS rate for NCI high-risk children and adolescents with BCR::ABL1-like ALL was 58% and 41%, respectively.[4 ] While it is more frequent in older and higher-risk patients, the BCR::ABL1-like subtype has also been identified in NCI standard-risk patients. In a COG study, 13.6% of 1,023 NCI standard-risk B-ALL patients were found to have BCR::ABL1-like ALL; these patients had an inferior EFS rate compared with non–BCR::ABL1-like standard-risk patients (82% vs. 91%), although no difference in OS rate (93% vs. 96%) was noted.[121 ] In one study of 40 BCR::ABL1-like patients, the adverse prognostic significance of this subtype appeared to be abrogated when patients were treated with risk-directed therapy on the basis of MRD levels.[122 ]The hallmark of BCR::ABL1-like ALL is activated kinase signaling, with approximately 35% to 50% containing CRLF2 genomic alterations [
1 ,118 ,123 ] and half of those cases containing concomitant JAK variants.[124 ]Many of the remaining cases of BCR::ABL1-like ALL have been noted to have a series of translocations involving tyrosine-kinase encoding ABL-class fusion genes, including ABL1, ABL2, CSF1R, and PDGFRB.[
4 ,119 ,125 ] Fusion proteins from these gene combinations have been noted in some cases to be transformative and have responded to tyrosine kinase inhibitors both in vitro and in vivo,[119 ] suggesting potential therapeutic strategies for these patients.BCR::ABL1-like ALL cases with non-CRLF2 genomic alterations represent approximately 3% of NCI standard-risk and 8% of NCI high-risk pediatric B-ALL cases.[
1 ] In a retrospective study of 122 pediatric patients (aged 1–18 years) with ABL-class fusions (all treated without tyrosine kinase inhibitors), the 5-year EFS rate was 59%, and the OS rate was 76%.[126 ]Approximately 9% of BCR::ABL1-like ALL cases result from rearrangements that lead to overexpression of a truncated erythropoietin receptor (EPOR).[
127 ] The C-terminal region of the receptor that is lost is the region that is altered in primary familial congenital polycythemia and that controls stability of the EPOR. The portion of the EPOR remaining is sufficient for JAK-STAT activation and for driving leukemia development. Point variants in kinase genes, aside from those in JAK1 and JAK2, are uncommon in patients with BCR::ABL1-like ALL.[8 ]CRLF2. Genomic alterations in CRLF2, a cytokine receptor gene located on the pseudoautosomal regions of the sex chromosomes, have been identified in 5% to 10% of cases of B-ALL. These alterations represent approximately 50% of cases of BCR::ABL1-like ALL.[
128 ,129 ,130 ] The chromosomal abnormalities that commonly lead to CRLF2 overexpression include translocations of the IGH locus (chromosome 14) to CRLF2 and interstitial deletions in pseudoautosomal regions of the sex chromosomes, resulting in a P2RY8::CRLF2 fusion.[8 ,123 ,128 ,129 ] These two genomic alterations are associated with distinctive clinical and biological characteristics.BCR::ABL1-like B-ALL with CRLF2 genomic alterations is observed in approximately 2% of NCI standard-risk and 5% of NCI high-risk pediatric B-ALL cases.[
1 ]ALL with genomic alterations in CRLF2 occurs at a higher incidence in children with Hispanic or Latino genetic ancestry [
123 ,131 ] and American Indian genetic ancestry.[85 ] In a study of 205 children with high-risk B-ALL, 18 of 51 (35.3%) Hispanic or Latino patients had CRLF2 rearrangements, compared with 11 of 154 (7.1%) cases of other declared ethnicity.[123 ] In a second study, only the frequency of IGH::CRLF2 fusions was increased in Hispanic or Latino children compared with non-Hispanic or non-Latino children with B-ALL (12% vs. 2.7%).[131 ] In this study, the percentage of B-ALL with P2RY8::CRLF2 fusions was approximately 6% and was not affected by ethnicity.The P2RY8::CRLF2 fusion is observed in 70% to 75% of pediatric patients with CRLF2 genomic alterations, and it occurs in younger patients (median age, approximately 4 years vs. 14 years for patients with IGH::CRLF2).[
132 ,133 ]P2RY8::CRLF2 occurs not infrequently with established chromosomal abnormalities (e.g., hyperdiploidy, iAMP21, dic(9;20)), while IGH::CRLF2 is generally mutually exclusive with known cytogenetic subgroups. CRLF2 genomic alterations are observed in approximately 60% of patients with Down syndrome and ALL, with P2RY8::CRLF2 fusions being more common than IGH::CRLF2 (approximately 80%–85% vs. 15%–20%).[129 ,132 ]IGH::CRLF2 and P2RY8::CRLF2 commonly occur as an early event in B-ALL development and show clonal prevalence.[
134 ] However, in some cases they appear to be a late event and show subclonal prevalence.[134 ] Loss of the CRLF2 genomic abnormality in some cases at relapse confirms the subclonal nature of the alteration in these cases.[132 ,135 ]CRLF2 abnormalities are strongly associated with the presence of IKZF1 deletions. Deletions of IKZF1 are more common in cases with IGH::CRLF2 fusions than in cases with P2RY8::CRLF2 fusions.[
133 ] Other recurring genomic alterations found in association with CRLF2 alterations include deletions in genes associated with B-cell differentiation (e.g., PAX5, BTG1, EBF1, etc.) and cell cycle control (CDKN2A), as well as genomic alterations activating JAK-STAT pathway signaling (e.g., IL7R and JAK variants).[4 ,123 ,124 ,129 ,136 ]Although the results of several retrospective studies suggest that CRLF2 abnormalities may have adverse prognostic significance in univariate analyses, most do not find this abnormality to be an independent predictor of outcome.[
123 ,128 ,129 ,137 ,138 ] For example, in a large European study, increased expression of CRLF2 was not associated with unfavorable outcome in multivariate analysis, while IKZF1 deletion and BCR::ABL1-like expression signatures were associated with unfavorable outcome.[120 ] Controversy exists about whether the prognostic significance of CRLF2 abnormalities should be analyzed on the basis of CRLF2 overexpression or on the presence of CRLF2 genomic alterations.[137 ,138 ] - IKZF1 deletions.
IKZF1 deletions, including deletions of the entire gene and deletions of specific exons, are present in approximately 15% of B-ALL cases. Less commonly, IKZF1 can be inactivated by deleterious point variants.[
117 ]Cases with IKZF1 deletions tend to occur in older children, have a higher WBC count at diagnosis, and are therefore more common in NCI high-risk patients than in NCI standard-risk patients.[
2 ,117 ,136 ,139 ,140 ] A high proportion of BCR::ABL1-positive cases have a deletion of IKZF1,[3 ,136 ] and ALL arising in children with Down syndrome appears to have elevated rates of IKZF1 deletions.[141 ]IKZF1 deletions are also common in cases with CRLF2 genomic alterations and in BCR::ABL1-like ALL cases.[87 ,116 ,136 ]Multiple reports have documented the adverse prognostic significance of an IKZF1 deletion, and most studies have reported that this deletion is an independent predictor of poor outcome in multivariate analyses.[
87 ,116 ,117 ,120 ,136 ,142 ,143 ,144 ,145 ,146 ,147 ,148 ,149 ]; [150 ][Level of evidence B4] However, the prognostic significance of IKZF1 may not apply equally across ALL biological subtypes, as illustrated by the apparent lack of prognostic significance in patients with ERG deletions.[87 ,88 ,89 ] Similarly, the prognostic significance of the IKZF1 deletion also appeared to be minimized in a cohort of COG patients with DUX4-rearranged ALL and with ERG transcriptional dysregulation that frequently occurred by ERG deletion.[6 ] The Associazione Italiana di Ematologia e Oncologia Pediatrica–Berlin-Frankfurt-Münster group reported that IKZF1 deletions were significant adverse prognostic factors only in B-ALL patients with high end-induction MRD and in whom co-occurrence of deletions of CDKN2A, CDKN2B, PAX5, or PAR1 (in the absence of ERG deletion) were identified.[151 ] The poor prognosis associated with IKZF1 alterations appears to be enhanced by the concomitant finding of deletion of 22q11.22. In a study of 1,310 patients with B-ALL, approximately one-half of the patients with IKZF1 alterations also had deletion of 22q11.22. The 5-year EFS rate was 43.3% for those with both abnormalities, compared with 68.5% for patients with IKZF1 alterations and wild-type 22q11.22 (P < .001).[152 ]There are few published results of changing therapy on the basis of IKZF1 gene status. The Malaysia-Singapore group published results of two consecutive trials. In the first trial (MS2003), IKZF1 status was not considered in risk stratification, while in the subsequent trial (MS2010), IKZF1-deleted patients were excluded from the standard-risk group. Thus, more IKZF1-deleted patients in the MS2010 trial received intensified therapy. Patients with IKZF1-deleted ALL had improved outcomes in MS2010 compared with patients in MS2003, but interpretation of this observation is limited by other changes in risk stratification and therapeutic differences between the two trials.[
153 ][Level of evidence B4]In the Dutch ALL11 study, patients with IKZF1 deletions had maintenance therapy extended by 1 year, with the goal of improving outcomes.[
154 ] The landmark analysis demonstrated an almost threefold reduction in relapse rate and an improvement in the 2-year EFS rate (from 74.4% to 91.2%), compared with historical controls. - MYC-rearranged ALL (8q24).
MYC gene rearrangements are a rare but recurrent finding in pediatric patients with B-ALL. Patients with rearrangements of the MYC gene and the IGH2, IGK, and IGL genes at 14q32, 2p12, and 22q11.2, respectively, have been reported.[
155 ,156 ,157 ] The lymphoblasts typically exhibit a precursor B-cell immunophenotype, with a French-American-British (FAB) L2 or L3 morphology, with no expression of surface immunoglobulin and kappa or lambda light chains. Concurrent MYC gene rearrangements have been observed along with additional cytogenetic rearrangements such as IGH::BCL2 or KMT2A.[157 ] Patients reported in the literature have been variably treated with ALL therapy or with mature B leukemia/lymphoma treatment protocols, and the optimal treatment for this patient group remains uncertain.[157 ]
- ETV6::RUNX1 fusion (t(12;21)(p13.2;q22.1)).
T-ALL cytogenetics/genomics
T-ALL is characterized by genomic alterations leading to activation of transcriptional programs related to T-cell development and by a high frequency of cases (approximately 60%) with variants in NOTCH1 and/or FBXW7 that result in activation of the NOTCH1 pathway.[
In Figure 5 below, pediatric T-ALL cases are divided into 10 molecular subtypes based on their RNA expression and gene variant status. These cases were derived from patients enrolled in SJCRH and COG clinical trials.[
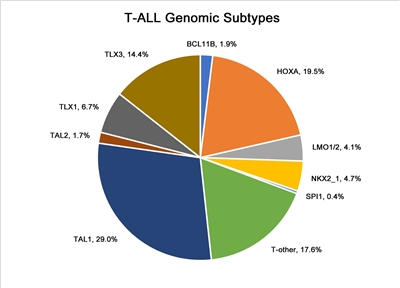
Figure 5. Genomic subtypes of T-ALL. The figure represents data from 466 children, adolescents, and young adults diagnosed with T-ALL and enrolled in St. Jude Children's Research Hospital or Children's Oncology Group clinical trials. Adapted from Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nature Genetics 54: 1376-1389, 2022.
- Notch pathway signaling.
Notch pathway signaling is commonly activated by NOTCH1 and FBXW7 gene variants in T-ALL, and these are the most commonly altered genes in pediatric T-ALL.[
158 ,163 ]NOTCH1-activating gene variants occur in approximately 50% to 60% of T-ALL cases, and FBXW7-inactivating gene variants occur in approximately 15% of cases. Approximately 60% of T-ALL cases have Notch pathway activation by variants in at least one of these genes.[164 ,165 ]The prognostic significance of NOTCH1 and FBXW7 variants may be modulated by genomic alterations in RAS and PTEN. The French Acute Lymphoblastic Leukaemia Study Group (FRALLE) and the Group for Research on Adult Acute Lymphoblastic Leukemia reported that patients having altered NOTCH1 or FBXW7 and wild-type PTEN and RAS constituted a favorable-risk group (i.e., low-risk group), while patients with PTEN or RAS variants, regardless of NOTCH1 and FBXW7 status, have a significantly higher risk of treatment failure (i.e., high-risk group).[
166 ,167 ] In the FRALLE study, the 5-year disease-free survival rate was 88% for the genetic low-risk group of patients and 60% for the genetic high-risk group of patients.[166 ] However, using the same criteria to define the genetic risk group, the Dana-Farber Cancer Institute consortium was unable to replicate these results. They reported a 5-year EFS rate of 86% for genetic low-risk patients and 79% for the genetic high-risk patients, a difference that was not statistically significant (P = .26).[165 ] - Chromosomal translocations.
Multiple chromosomal translocations have been identified in T-ALL that lead to deregulated expression of the target genes. These chromosome rearrangements fuse genes encoding transcription factors (e.g., TAL1, TAL2, LMO1, LMO2, LYL1, TLX1, TLX3, NKX2-I, HOXA, and MYB) to one of the T-cell receptor loci (or to other genes) and result in deregulated expression of these transcription factors in leukemia cells.[
158 ,159 ,168 ,169 ,170 ,171 ,172 ] These translocations are often not apparent by examining a standard karyotype, but can be identified using more sensitive screening techniques, including FISH or PCR.[159 ] Variants in a noncoding region near the TAL1 gene that produce a super-enhancer upstream of TAL1 represent nontranslocation genomic alterations that can also activate TAL1 transcription to induce T-ALL.[161 ]Translocations resulting in chimeric fusion proteins are also observed in T-ALL.[
166 ]- A NUP214::ABL1 fusion has been noted in 4% to 6% of T-ALL cases and is observed in both adults and children, with a male predominance.[
173 ,174 ,175 ] The fusion is cytogenetically cryptic and is seen in FISH on amplified episomes or, more rarely, as a small homogeneous staining region.[175 ] T-ALL may also uncommonly show ABL1 fusion proteins with other gene partners (e.g., ETV6, BCR, and EML1).[175 ] ABL tyrosine kinase inhibitors, such as imatinib or dasatinib, may demonstrate therapeutic benefits in this T-ALL subtype,[173 ,174 ,176 ] although clinical experience with this strategy is very limited.[177 ,178 ,179 ] - Gene fusions involving SPI1 (encoding the transcription factor PU.1) were reported in 4% of Japanese children with T-ALL.[
180 ] Fusion partners included STMN1 and TCF7. T-ALL cases with SPI1 fusions had a particularly poor prognosis; six of seven affected individuals died within 3 years of diagnosis of early relapse. - BCL11B is a zinc finger transcription factor that plays a dual role as a transcription activator and repressor. It is known to play a critical role in T-cell differentiation. In T-ALL, the BCL11B gene is involved in a t(5;14)(q35;q32) translocation where a distal BCL11B enhancer drives aberrant expression of TLX3 (or NKX2-5).[
181 ] In the process of donating its enhancer, one allele of BCL11B is inactivated. However, the resulting haploinsufficient state itself may also play a role in tumor pathogenesis. The role of BCL11B as a tumor suppressor gene is supported by the finding that about 16% of patients have T-ALL that harbors deletions or missense variants.[158 ,182 ] As described in the sections for early T-cell precursor (ETP) and T/myeloid mixed phenotype acute leukemia (T/M MPAL), BCL11B may also be leukemogenic through overexpression. - Other recurring gene fusions in T-ALL patients include those involving MLLT10, KMT2A, NUP214, and NUP98.[
158 ,162 ]
- A NUP214::ABL1 fusion has been noted in 4% to 6% of T-ALL cases and is observed in both adults and children, with a male predominance.[
- Ploidy.
- Recurrent abnormalities in chromosome number are much less common in T-ALL than in B-ALL. One study included 2,250 pediatric patients with T-ALL who were treated in Associazione Italiana di Ematologia e Oncologia Pediatrica/Berlin-Frankfurt-Münster protocols. The study found that near tetraploidy (DNA index, 1.79–2.28 or 81–103 chromosomes), observed in 1.4% of patients, was associated with favorable disease features and outcomes.[
183 ]
- Recurrent abnormalities in chromosome number are much less common in T-ALL than in B-ALL. One study included 2,250 pediatric patients with T-ALL who were treated in Associazione Italiana di Ematologia e Oncologia Pediatrica/Berlin-Frankfurt-Münster protocols. The study found that near tetraploidy (DNA index, 1.79–2.28 or 81–103 chromosomes), observed in 1.4% of patients, was associated with favorable disease features and outcomes.[
Early T-cell precursor (ETP) ALL cytogenetics/genomics
Detailed molecular characterization of ETP ALL showed this entity to be highly heterogeneous at the molecular level, with no single gene affected by variant or copy number alteration in more than one-third of cases.[
Studies have found that the absence of biallelic deletion of the TCR-gamma locus (ABD), as detected by comparative genomic hybridization and/or quantitative DNA-PCR, was associated with early treatment failure in patients with T-ALL.[
Allele-specific, generally high expression of BCL11B plays an oncogenic role in a subset of cases identified as ETP ALL (7 of 58 in one study) as well as in up to 30% to 40% of lineage ambiguous leukemia T/M mixed phenotype acute leukemia (T/M MPAL).[
- One such alteration is t(2;14)(q22;q32), which produces an in-frame ZEB2::BCL11B fusion gene.
- Other structural variants leading to allele-specific deregulated BCL11B expression include structural variants that juxtapose regulatory sequences of active genes (e.g., ARID1B [chromosome 6], BENC [chromosome 7], and CDK6 [chromosome 7]) upstream or downstream of the BCL11B locus leading to aberrant expression in a process called enhancer hijacking.
- Finally, in about 20% of cases with deregulated BCL11B expression, a translocation cannot be identified. In many such cases, amplification of a downstream enhancer, BCL11B enhancer tandem amplification (BETA), leads to BCL11B promoter driven transcription.
- There is a high prevalence of FLT3 alterations and JAK/STAT activation in acute leukemias driven by genomic alterations leading to BCL11B expression.[
187 ]
Mixed phenotype acute leukemia (MPAL) cytogenetics/genomics
For acute leukemias of ambiguous lineage, the WHO classification system is summarized in Table 3.[
| Condition | Definition |
|---|---|
| MPAL = mixed phenotype acute leukemia; NOS = not otherwise specified. | |
| a Adapted from Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.[ |
|
| Acute undifferentiated leukemia | Acute leukemia that does not express any marker considered specific for either lymphoid or myeloid lineage |
| MPAL withBCR::ABL1(t(9;22)(q34;q11.2)) | Acute leukemia meeting the diagnostic criteria for MPAL in which the blasts also have the (9;22) translocation or theBCR::ABL1rearrangement |
| MPAL withKMT2A(t(v;11q23)) | Acute leukemia meeting the diagnostic criteria for MPAL in which the blasts also have a translocation involving theKMT2Agene |
| MPAL, B/myeloid, NOS (B/M MPAL) | Acute leukemia meeting the diagnostic criteria for assignment to both B and myeloid lineage, in which the blasts lack genetic abnormalities involvingBCR::ABL1orKMT2A |
| MPAL, T/myeloid, NOS (T/M MPAL) | Acute leukemia meeting the diagnostic criteria for assignment to both T and myeloid lineage, in which the blasts lack genetic abnormalities involvingBCR::ABL1orKMT2A |
| MPAL, B/myeloid, NOS—rare types | Acute leukemia meeting the diagnostic criteria for assignment to both B and T lineage |
| Other ambiguous lineage leukemias | Natural killer–cell lymphoblastic leukemia/lymphoma |
| Lineage | Criteria |
|---|---|
| a Adapted from Arber et al.[ |
|
| b Strong defined as equal to or brighter than the normal B or T cells in the sample. | |
| Myeloid lineage | Myeloperoxidase (flow cytometry, immunohistochemistry, or cytochemistry);or monocytic differentiation (at least two of the following: nonspecific esterase cytochemistry, CD11c, CD14, CD64, lysozyme) |
| T lineage | Strongb cytoplasmic CD3 (with antibodies to CD3 epsilon chain);or surface CD3 |
| B lineage | Strongb CD19 with at least one of the following strongly expressed: CD79a, cytoplasmic CD22, or CD10;or weak CD19 with at least two of the following strongly expressed: CD79a, cytoplasmic CD22, or CD10 |
The classification system for MPAL includes two entities that are defined by their primary molecular alteration: MPAL with BCR::ABL1 translocation and MPAL with KMT2A rearrangement. The genomic alterations associated with the MPAL, B/myeloid, NOS (B/M MPAL) and MPAL, T/myeloid, NOS (T/M MPAL) entities are distinctive, as described below:
- B/M MPAL.
- Among 115 MPAL cases for which genomic characterization was performed, 35 (30%) were B/M MPAL. There were an additional 16 MPAL cases (14%) with KMT2A rearrangements, 15 of whom showed a B/myeloid immunophenotype.
- Approximately one-half of B/M MPAL cases had rearrangements of ZNF384 with recurrent fusion partners, including TCF3 and EP300. These cases had gene expression profiles indistinguishable from B-ALL cases with ZNF384 rearrangements.[
100 ] - Approximately two-thirds of B/M MPAL cases had RAS pathway alterations, with NRAS and PTPN11 being the most commonly altered genes.[
100 ] - Genes encoding epigenetic regulators (e.g., MLLT3, KDM6A, EP300, and CREBBP) are altered in approximately two-thirds of B/M MPAL cases.[
100 ]
- T/M MPAL.
- Among 115 MPAL cases for which genomic characterization was performed, 49 (43%) were T/M MPAL.[
100 ] The genomic features of the T/M MPAL cases shared commonalities with those of ETP ALL, suggesting that T/M MPAL and ETP ALL are similar entities along the spectrum of immature leukemias. - Compared with T-ALL, T/M MPAL showed a lower rate of alterations in the core T-ALL transcription factors (TAL1, TAL2, TLX1, TLX3, LMO1, LMO2, NKX2-1, HOXA10, and LYL1) (63% vs. 16%, respectively).[
100 ] A similar lower rate was also observed for ETP ALL. - CDKN2A, CDKN2B, and NOTCH1 variants, which are present in approximately two-thirds of T-ALL cases, were much less common in T/M MPAL cases. By contrast, WT1 variants occurred in approximately 40% of T/M MPAL, but in less than 10% of T-ALL cases.[
100 ] - One-third of T/M MPAL cases have genomic alterations associated with BCL11B that lead to allele-specific, generally high expression of BCL11B.[
187 ,188 ]- One such alteration is t(2;14)(q22;q32), which produces an in-frame ZEB2::BCL11B fusion gene that leads to deregulated expression of BCL11B.
- Other alterations leading to allele-specific deregulated BCL11B expression include structural variants that juxtapose regulatory sequences of active genes (e.g., ARID1B [chromosome 6], BENC [chromosome 7], and CDK6 [chromosome 7]) upstream or downstream of the BCL11B locus in a process called enhancer hijacking.
- Finally, a translocation cannot be identified in about 20% of cases with deregulated BCL11B overexpression. In such cases, amplification of a downstream enhancer, BCL11B enhancer tandem amplification (BETA), leads to BCL11B promoter driven transcription.
- There is a high prevalence of FLT3 alterations and JAK/STAT activation in acute leukemias driven by genomic alterations leading to BCL11B overexpression.
- RAS and JAK-STAT pathway variants were common in the T/M MPAL and ETP ALL cases, while the PI3K signaling pathway is more commonly altered in T-ALL.[
100 ] For T/M MPAL, the most commonly altered signaling pathway gene was FLT3 (43% of cases). FLT3 variants tended to be mutually exclusive with RAS pathway variants. - Genes encoding epigenetic regulators (e.g., EZH2 and PHF6) were altered in approximately two-thirds of T/M MPAL cases.[
100 ]
- Among 115 MPAL cases for which genomic characterization was performed, 49 (43%) were T/M MPAL.[
Gene polymorphisms in drug metabolic pathways
Several polymorphisms of genes involved in the metabolism of chemotherapeutic agents have been reported to have prognostic significance in childhood ALL.[
- TPMT.
Patients with variant phenotypes of TPMT (a gene involved in the metabolism of thiopurines such as mercaptopurine) appear to have more favorable outcomes,[
194 ] although such patients may also be at higher risk of developing significant treatment-related toxicities, including myelosuppression, infection, and second malignancies.[195 ,196 ] Patients with homozygosity for TPMT variants associated with low enzymatic activity tolerate only very low doses of mercaptopurine (approximately 10% of the standard dose) and are treated with reduced doses of mercaptopurine to avoid excessive toxicity. Patients who are heterozygous for this variant enzyme gene generally tolerate mercaptopurine without serious toxicity, but they do require more frequent dose reductions for hematologic toxicity than do patients who are homozygous for the normal allele.[197 ,198 ] - NUDT15.
Germline variants in NUDT15 that reduce or abolish activity of this enzyme also lead to diminished tolerance to thiopurines.[
197 ,199 ] The NUDT15 variants are most common in East Asian and Hispanic patients, and they are rare in European and African patients. Patients homozygous for the risk variants tolerate only very low doses of mercaptopurine, while patients heterozygous for the risk alleles tolerate lower doses than do patients homozygous for the wild-type allele (approximately 25% dose reduction on average), but there is broad overlap in tolerated doses between the two groups.[197 ,200 ] - CEP72.
Gene polymorphisms may also affect the expression of proteins that play central roles in the cellular effects of anticancer drugs. As an example, patients who are homozygous for a polymorphism in the promoter region of CEP72 (a centrosomal protein involved in microtubule formation) are at increased risk of vincristine neurotoxicity.[
201 ] - Single nucleotide polymorphisms.
Genome-wide polymorphism analysis has identified specific single nucleotide polymorphisms associated with high end-induction MRD and risk of relapse. Polymorphisms of interleukin-15, as well as genes associated with the metabolism of etoposide and methotrexate, were significantly associated with treatment response in two large cohorts of ALL patients treated on SJCRH and COG protocols.[
202 ] Polymorphic variants involving the reduced folate carrier and methotrexate metabolism have been linked to toxicity and outcome.[203 ,204 ] While these associations suggest that individual variations in drug metabolism can affect outcome, few studies have attempted to adjust for these variations. It is unknown whether individualized dose modification on the basis of these findings will improve outcomes.
References:
- Brady SW, Roberts KG, Gu Z, et al.: The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet 54 (9): 1376-1389, 2022.
- Mullighan CG, Goorha S, Radtke I, et al.: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 (7137): 758-64, 2007.
- Mullighan CG, Miller CB, Radtke I, et al.: BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 (7191): 110-4, 2008.
- Roberts KG, Li Y, Payne-Turner D, et al.: Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 371 (11): 1005-15, 2014.
- Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, et al.: Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun 7: 11790, 2016.
- Zhang J, McCastlain K, Yoshihara H, et al.: Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48 (12): 1481-1489, 2016.
- Holmfeldt L, Wei L, Diaz-Flores E, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 45 (3): 242-52, 2013.
- Loh ML, Zhang J, Harvey RC, et al.: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 121 (3): 485-8, 2013.
- Bercovich D, Ganmore I, Scott LM, et al.: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 372 (9648): 1484-92, 2008.
- Li Z, Chang TC, Junco JJ, et al.: Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood 142 (2): 172-184, 2023.
- Andersson AK, Ma J, Wang J, et al.: The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet 47 (4): 330-7, 2015.
- Ma X, Edmonson M, Yergeau D, et al.: Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 6: 6604, 2015.
- Meyer JA, Wang J, Hogan LE, et al.: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 45 (3): 290-4, 2013.
- Li B, Li H, Bai Y, et al.: Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med 21 (6): 563-71, 2015.
- Mullighan CG, Zhang J, Kasper LH, et al.: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 471 (7337): 235-9, 2011.
- Mattano LA, Devidas M, Maloney KW, et al.: Favorable Trisomies and ETV6-RUNX1 Predict Cure in Low-Risk B-Cell Acute Lymphoblastic Leukemia: Results From Children's Oncology Group Trial AALL0331. J Clin Oncol 39 (14): 1540-1552, 2021.
- Moorman AV, Ensor HM, Richards SM, et al.: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 11 (5): 429-38, 2010.
- Alaggio R, Amador C, Anagnostopoulos I, et al.: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 36 (7): 1720-1748, 2022.
- Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 48 (8): 637-60, 2009.
- Aricò M, Valsecchi MG, Rizzari C, et al.: Long-term results of the AIEOP-ALL-95 Trial for Childhood Acute Lymphoblastic Leukemia: insight on the prognostic value of DNA index in the framework of Berlin-Frankfurt-Muenster based chemotherapy. J Clin Oncol 26 (2): 283-9, 2008.
- Dastugue N, Suciu S, Plat G, et al.: Hyperdiploidy with 58-66 chromosomes in childhood B-acute lymphoblastic leukemia is highly curable: 58951 CLG-EORTC results. Blood 121 (13): 2415-23, 2013.
- Synold TW, Relling MV, Boyett JM, et al.: Blast cell methotrexate-polyglutamate accumulation in vivo differs by lineage, ploidy, and methotrexate dose in acute lymphoblastic leukemia. J Clin Invest 94 (5): 1996-2001, 1994.
- Moorman AV, Richards SM, Martineau M, et al.: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 102 (8): 2756-62, 2003.
- Chilton L, Buck G, Harrison CJ, et al.: High hyperdiploidy among adolescents and adults with acute lymphoblastic leukaemia (ALL): cytogenetic features, clinical characteristics and outcome. Leukemia 28 (7): 1511-8, 2014.
- Sutcliffe MJ, Shuster JJ, Sather HN, et al.: High concordance from independent studies by the Children's Cancer Group (CCG) and Pediatric Oncology Group (POG) associating favorable prognosis with combined trisomies 4, 10, and 17 in children with NCI Standard-Risk B-precursor Acute Lymphoblastic Leukemia: a Children's Oncology Group (COG) initiative. Leukemia 19 (5): 734-40, 2005.
- Harris MB, Shuster JJ, Carroll A, et al.: Trisomy of leukemic cell chromosomes 4 and 10 identifies children with B-progenitor cell acute lymphoblastic leukemia with a very low risk of treatment failure: a Pediatric Oncology Group study. Blood 79 (12): 3316-24, 1992.
- Enshaei A, Vora A, Harrison CJ, et al.: Defining low-risk high hyperdiploidy in patients with paediatric acute lymphoblastic leukaemia: a retrospective analysis of data from the UKALL97/99 and UKALL2003 clinical trials. Lancet Haematol 8 (11): e828-e839, 2021.
- Heerema NA, Harbott J, Galimberti S, et al.: Secondary cytogenetic aberrations in childhood Philadelphia chromosome positive acute lymphoblastic leukemia are nonrandom and may be associated with outcome. Leukemia 18 (4): 693-702, 2004.
- Carroll AJ, Shago M, Mikhail FM, et al.: Masked hypodiploidy: Hypodiploid acute lymphoblastic leukemia (ALL) mimicking hyperdiploid ALL in children: A report from the Children's Oncology Group. Cancer Genet 238: 62-68, 2019.
- Nachman JB, Heerema NA, Sather H, et al.: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 110 (4): 1112-5, 2007.
- Raimondi SC, Zhou Y, Shurtleff SA, et al.: Near-triploidy and near-tetraploidy in childhood acute lymphoblastic leukemia: association with B-lineage blast cells carrying the ETV6-RUNX1 fusion, T-lineage immunophenotype, and favorable outcome. Cancer Genet Cytogenet 169 (1): 50-7, 2006.
- Attarbaschi A, Mann G, König M, et al.: Incidence and relevance of secondary chromosome abnormalities in childhood TEL/AML1+ acute lymphoblastic leukemia: an interphase FISH analysis. Leukemia 18 (10): 1611-6, 2004.
- Lemez P, Attarbaschi A, Béné MC, et al.: Childhood near-tetraploid acute lymphoblastic leukemia: an EGIL study on 36 cases. Eur J Haematol 85 (4): 300-8, 2010.
- Paulsson K, Lilljebjörn H, Biloglav A, et al.: The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet 47 (6): 672-6, 2015.
- Harrison CJ, Moorman AV, Broadfield ZJ, et al.: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 125 (5): 552-9, 2004.
- Mullighan CG, Jeha S, Pei D, et al.: Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 126 (26): 2896-9, 2015.
- Pui CH, Rebora P, Schrappe M, et al.: Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J Clin Oncol 37 (10): 770-779, 2019.
- McNeer JL, Devidas M, Dai Y, et al.: Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group. J Clin Oncol 37 (10): 780-789, 2019.
- Irving J, Matheson E, Minto L, et al.: Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood 124 (23): 3420-30, 2014.
- Qian M, Cao X, Devidas M, et al.: TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J Clin Oncol 36 (6): 591-599, 2018.
- Rubnitz JE, Wichlan D, Devidas M, et al.: Prospective analysis of TEL gene rearrangements in childhood acute lymphoblastic leukemia: a Children's Oncology Group study. J Clin Oncol 26 (13): 2186-91, 2008.
- Kanerva J, Saarinen-Pihkala UM, Niini T, et al.: Favorable outcome in 20-year follow-up of children with very-low-risk ALL and minimal standard therapy, with special reference to TEL-AML1 fusion. Pediatr Blood Cancer 42 (1): 30-5, 2004.
- Aldrich MC, Zhang L, Wiemels JL, et al.: Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer Epidemiol Biomarkers Prev 15 (3): 578-81, 2006.
- Loh ML, Goldwasser MA, Silverman LB, et al.: Prospective analysis of TEL/AML1-positive patients treated on Dana-Farber Cancer Institute Consortium Protocol 95-01. Blood 107 (11): 4508-13, 2006.
- Borowitz MJ, Devidas M, Hunger SP, et al.: Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood 111 (12): 5477-85, 2008.
- Madzo J, Zuna J, Muzíková K, et al.: Slower molecular response to treatment predicts poor outcome in patients with TEL/AML1 positive acute lymphoblastic leukemia: prospective real-time quantitative reverse transcriptase-polymerase chain reaction study. Cancer 97 (1): 105-13, 2003.
- Bhojwani D, Pei D, Sandlund JT, et al.: ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 26 (2): 265-70, 2012.
- Enshaei A, Schwab CJ, Konn ZJ, et al.: Long-term follow-up of ETV6-RUNX1 ALL reveals that NCI risk, rather than secondary genetic abnormalities, is the key risk factor. Leukemia 27 (11): 2256-9, 2013.
- Barbany G, Andersen MK, Autio K, et al.: Additional aberrations of the ETV6 and RUNX1 genes have no prognostic impact in 229 t(12;21)(p13;q22)-positive B-cell precursor acute lymphoblastic leukaemias treated according to the NOPHO-ALL-2000 protocol. Leuk Res 36 (7): 936-8, 2012.
- Forestier E, Heyman M, Andersen MK, et al.: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 140 (6): 665-72, 2008.
- Seeger K, Stackelberg AV, Taube T, et al.: Relapse of TEL-AML1--positive acute lymphoblastic leukemia in childhood: a matched-pair analysis. J Clin Oncol 19 (13): 3188-93, 2001.
- Gandemer V, Chevret S, Petit A, et al.: Excellent prognosis of late relapses of ETV6/RUNX1-positive childhood acute lymphoblastic leukemia: lessons from the FRALLE 93 protocol. Haematologica 97 (11): 1743-50, 2012.
- Zuna J, Ford AM, Peham M, et al.: TEL deletion analysis supports a novel view of relapse in childhood acute lymphoblastic leukemia. Clin Cancer Res 10 (16): 5355-60, 2004.
- van Delft FW, Horsley S, Colman S, et al.: Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 (23): 6247-54, 2011.
- Aricò M, Schrappe M, Hunger SP, et al.: Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol 28 (31): 4755-61, 2010.
- Schrappe M, Aricò M, Harbott J, et al.: Philadelphia chromosome-positive (Ph+) childhood acute lymphoblastic leukemia: good initial steroid response allows early prediction of a favorable treatment outcome. Blood 92 (8): 2730-41, 1998.
- Ribeiro RC, Broniscer A, Rivera GK, et al.: Philadelphia chromosome-positive acute lymphoblastic leukemia in children: durable responses to chemotherapy associated with low initial white blood cell counts. Leukemia 11 (9): 1493-6, 1997.
- Biondi A, Schrappe M, De Lorenzo P, et al.: Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol 13 (9): 936-45, 2012.
- Schultz KR, Bowman WP, Aledo A, et al.: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27 (31): 5175-81, 2009.
- Schultz KR, Carroll A, Heerema NA, et al.: Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children's Oncology Group study AALL0031. Leukemia 28 (7): 1467-71, 2014.
- Duffield AS, Mullighan CG, Borowitz MJ: International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch 482 (1): 11-26, 2023.
- Short NJ, Jabbour E, Macaron W, et al.: Ultrasensitive NGS MRD assessment in Ph+ ALL: Prognostic impact and correlation with RT-PCR for BCR::ABL1. Am J Hematol 98 (8): 1196-1203, 2023.
- Hovorkova L, Zaliova M, Venn NC, et al.: Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood 129 (20): 2771-2781, 2017.
- Zuna J, Hovorkova L, Krotka J, et al.: Minimal residual disease in BCR::ABL1-positive acute lymphoblastic leukemia: different significance in typical ALL and in CML-like disease. Leukemia 36 (12): 2793-2801, 2022.
- Hunger SP, Tran TH, Saha V, et al.: Dasatinib with intensive chemotherapy in de novo paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (CA180-372/COG AALL1122): a single-arm, multicentre, phase 2 trial. Lancet Haematol 10 (7): e510-e520, 2023.
- Pui CH, Chessells JM, Camitta B, et al.: Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 17 (4): 700-6, 2003.
- Johansson B, Moorman AV, Haas OA, et al.: Hematologic malignancies with t(4;11)(q21;q23)--a cytogenetic, morphologic, immunophenotypic and clinical study of 183 cases. European 11q23 Workshop participants. Leukemia 12 (5): 779-87, 1998.
- Raimondi SC, Peiper SC, Kitchingman GR, et al.: Childhood acute lymphoblastic leukemia with chromosomal breakpoints at 11q23. Blood 73 (6): 1627-34, 1989.
- Harrison CJ, Moorman AV, Barber KE, et al.: Interphase molecular cytogenetic screening for chromosomal abnormalities of prognostic significance in childhood acute lymphoblastic leukaemia: a UK Cancer Cytogenetics Group Study. Br J Haematol 129 (4): 520-30, 2005.
- Pui CH, Pei D, Campana D, et al.: A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia 28 (12): 2336-43, 2014.
- Pieters R, Schrappe M, De Lorenzo P, et al.: A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370 (9583): 240-50, 2007.
- Attarbaschi A, Möricke A, Harrison CJ, et al.: Outcomes of Childhood Noninfant Acute Lymphoblastic Leukemia With 11q23/KMT2A Rearrangements in a Modern Therapy Era: A Retrospective International Study. J Clin Oncol 41 (7): 1404-1422, 2023.
- Isobe T, Takagi M, Sato-Otsubo A, et al.: Multi-omics analysis defines highly refractory RAS burdened immature subgroup of infant acute lymphoblastic leukemia. Nat Commun 13 (1): 4501, 2022.
- Pui CH, Gaynon PS, Boyett JM, et al.: Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 359 (9321): 1909-15, 2002.
- Rubnitz JE, Camitta BM, Mahmoud H, et al.: Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol 17 (1): 191-6, 1999.
- Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 87 (4): 1211-24, 1996.
- Uckun FM, Sensel MG, Sather HN, et al.: Clinical significance of translocation t(1;19) in childhood acute lymphoblastic leukemia in the context of contemporary therapies: a report from the Children's Cancer Group. J Clin Oncol 16 (2): 527-35, 1998.
- Fischer U, Forster M, Rinaldi A, et al.: Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47 (9): 1020-9, 2015.
- Pui CH, Sandlund JT, Pei D, et al.: Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 290 (15): 2001-7, 2003.
- Crist WM, Carroll AJ, Shuster JJ, et al.: Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): a Pediatric Oncology Group study. Blood 76 (1): 117-22, 1990.
- Andersen MK, Autio K, Barbany G, et al.: Paediatric B-cell precursor acute lymphoblastic leukaemia with t(1;19)(q23;p13): clinical and cytogenetic characteristics of 47 cases from the Nordic countries treated according to NOPHO protocols. Br J Haematol 155 (2): 235-43, 2011.
- Pui CH, Campana D, Pei D, et al.: Treating childhood acute lymphoblastic leukemia without cranial irradiation. N Engl J Med 360 (26): 2730-41, 2009.
- Jeha S, Pei D, Raimondi SC, et al.: Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 23 (8): 1406-9, 2009.
- Minson KA, Prasad P, Vear S, et al.: t(17;19) in Children with Acute Lymphocytic Leukemia: A Report of 3 Cases and a Review of the Literature. Case Rep Hematol 2013: 563291, 2013.
- Lee SHR, Antillon-Klussmann F, Pei D, et al.: Association of Genetic Ancestry With the Molecular Subtypes and Prognosis of Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 8 (3): 354-363, 2022.
- Zaliova M, Potuckova E, Hovorkova L, et al.: ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 104 (7): 1407-1416, 2019.
- Harvey RC, Mullighan CG, Wang X, et al.: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 (23): 4874-84, 2010.
- Zaliova M, Zimmermannova O, Dörge P, et al.: ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia 28 (1): 182-5, 2014.
- Clappier E, Auclerc MF, Rapion J, et al.: An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 28 (1): 70-7, 2014.
- Li Z, Lee SHR, Chin WHN, et al.: Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv 5 (23): 5226-5238, 2021.
- Gu Z, Churchman M, Roberts K, et al.: Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 7: 13331, 2016.
- Liu YF, Wang BY, Zhang WN, et al.: Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8: 173-83, 2016.
- Suzuki K, Okuno Y, Kawashima N, et al.: MEF2D-BCL9 Fusion Gene Is Associated With High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Adolescents. J Clin Oncol 34 (28): 3451-9, 2016.
- Lilljebjörn H, Ågerstam H, Orsmark-Pietras C, et al.: RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia 28 (4): 977-9, 2014.
- Hirabayashi S, Ohki K, Nakabayashi K, et al.: ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102 (1): 118-129, 2017.
- Qian M, Zhang H, Kham SK, et al.: Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res 27 (2): 185-195, 2017.
- Hirabayashi S, Butler ER, Ohki K, et al.: Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: a retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 35 (11): 3272-3277, 2021.
- Shago M, Abla O, Hitzler J, et al.: Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr Blood Cancer 63 (11): 1915-21, 2016.
- Yao L, Cen J, Pan J, et al.: TAF15-ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet 211: 1-4, 2017.
- Alexander TB, Gu Z, Iacobucci I, et al.: The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562 (7727): 373-379, 2018.
- Boer JM, Valsecchi MG, Hormann FM, et al.: Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 35 (10): 2978-2982, 2021.
- De Lorenzo P, Moorman AV, Pieters R, et al.: Cytogenetics and outcome of infants with acute lymphoblastic leukemia and absence of MLL rearrangements. Leukemia 28 (2): 428-30, 2014.
- Fazio G, Bardini M, De Lorenzo P, et al.: Recurrent genetic fusions redefine MLL germ line acute lymphoblastic leukemia in infants. Blood 137 (14): 1980-1984, 2021.
- Arber DA, Orazi A, Hasserjian R, et al.: The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127 (20): 2391-405, 2016.
- Hogan TF, Koss W, Murgo AJ, et al.: Acute lymphoblastic leukemia with chromosomal 5;14 translocation and hypereosinophilia: case report and literature review. J Clin Oncol 5 (3): 382-90, 1987.
- Grimaldi JC, Meeker TC: The t(5;14) chromosomal translocation in a case of acute lymphocytic leukemia joins the interleukin-3 gene to the immunoglobulin heavy chain gene. Blood 73 (8): 2081-5, 1989.
- Meeker TC, Hardy D, Willman C, et al.: Activation of the interleukin-3 gene by chromosome translocation in acute lymphocytic leukemia with eosinophilia. Blood 76 (2): 285-9, 1990.
- Sutton R, Lonergan M, Tapp H, et al.: Two cases of hypereosinophilia and high-risk acute lymphoblastic leukemia. Leukemia 22 (7): 1463-5, 2008.
- Koleilat A, Smadbeck JB, Zepeda-Mendoza CJ, et al.: Characterization of unusual iAMP21 B-lymphoblastic leukemia (iAMP21-ALL) from the Mayo Clinic and Children's Oncology Group. Genes Chromosomes Cancer 61 (12): 710-719, 2022.
- Heerema NA, Carroll AJ, Devidas M, et al.: Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children's oncology group studies: a report from the children's oncology group. J Clin Oncol 31 (27): 3397-402, 2013.
- Moorman AV, Robinson H, Schwab C, et al.: Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol 31 (27): 3389-96, 2013.
- Harrison CJ, Moorman AV, Schwab C, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 28 (5): 1015-21, 2014.
- Gu Z, Churchman ML, Roberts KG, et al.: PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 51 (2): 296-307, 2019.
- Strehl S, König M, Dworzak MN, et al.: PAX5/ETV6 fusion defines cytogenetic entity dic(9;12)(p13;p13). Leukemia 17 (6): 1121-3, 2003.
- Schwab C, Nebral K, Chilton L, et al.: Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv 1 (19): 1473-7, 2017.
- Den Boer ML, van Slegtenhorst M, De Menezes RX, et al.: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 10 (2): 125-34, 2009.
- Mullighan CG, Su X, Zhang J, et al.: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 360 (5): 470-80, 2009.
- Reshmi SC, Harvey RC, Roberts KG, et al.: Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood 129 (25): 3352-3361, 2017.
- Roberts KG, Morin RD, Zhang J, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22 (2): 153-66, 2012.
- van der Veer A, Waanders E, Pieters R, et al.: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 122 (15): 2622-9, 2013.
- Roberts KG, Reshmi SC, Harvey RC, et al.: Genomic and outcome analyses of Ph-like ALL in NCI standard-risk patients: a report from the Children's Oncology Group. Blood 132 (8): 815-824, 2018.
- Roberts KG, Pei D, Campana D, et al.: Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol 32 (27): 3012-20, 2014.
- Harvey RC, Mullighan CG, Chen IM, et al.: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 115 (26): 5312-21, 2010.
- Mullighan CG, Collins-Underwood JR, Phillips LA, et al.: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41 (11): 1243-6, 2009.
- Schwab C, Roberts K, Boer JM, et al.: SSBP2-CSF1R is a recurrent fusion in B-lineage acute lymphoblastic leukemia with diverse genetic presentation and variable outcome. Blood 137 (13): 1835-1838, 2021.
- den Boer ML, Cario G, Moorman AV, et al.: Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol 8 (1): e55-e66, 2021.
- Iacobucci I, Li Y, Roberts KG, et al.: Truncating Erythropoietin Receptor Rearrangements in Acute Lymphoblastic Leukemia. Cancer Cell 29 (2): 186-200, 2016.
- Cario G, Zimmermann M, Romey R, et al.: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 115 (26): 5393-7, 2010.
- Ensor HM, Schwab C, Russell LJ, et al.: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 117 (7): 2129-36, 2011.
- Schmäh J, Fedders B, Panzer-Grümayer R, et al.: Molecular characterization of acute lymphoblastic leukemia with high CRLF2 gene expression in childhood. Pediatr Blood Cancer 64 (10): , 2017.
- Raca G, Abdel-Azim H, Yue F, et al.: Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children-a novel health disparity. Leukemia 35 (8): 2399-2402, 2021.
- Vesely C, Frech C, Eckert C, et al.: Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia 31 (7): 1491-1501, 2017.
- Russell LJ, Jones L, Enshaei A, et al.: Characterisation of the genomic landscape of CRLF2-rearranged acute lymphoblastic leukemia. Genes Chromosomes Cancer 56 (5): 363-372, 2017.
- Potter N, Jones L, Blair H, et al.: Single-cell analysis identifies CRLF2 rearrangements as both early and late events in Down syndrome and non-Down syndrome acute lymphoblastic leukaemia. Leukemia 33 (4): 893-904, 2019.
- Morak M, Attarbaschi A, Fischer S, et al.: Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood 120 (26): 5134-42, 2012.
- Schwab CJ, Chilton L, Morrison H, et al.: Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98 (7): 1081-8, 2013.
- Chen IM, Harvey RC, Mullighan CG, et al.: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 119 (15): 3512-22, 2012.
- Palmi C, Vendramini E, Silvestri D, et al.: Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 26 (10): 2245-53, 2012.
- Clappier E, Grardel N, Bakkus M, et al.: IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children's Leukemia Group study 58951. Leukemia 29 (11): 2154-61, 2015.
- Srinivasan S, Ramanathan S, Kumar S, et al.: Prevalence and prognostic significance of IKZF1 deletion in paediatric acute lymphoblastic leukemia: A systematic review and meta-analysis. Ann Hematol 102 (8): 2165-2179, 2023.
- Buitenkamp TD, Pieters R, Gallimore NE, et al.: Outcome in children with Down's syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia 26 (10): 2204-11, 2012.
- Krentz S, Hof J, Mendioroz A, et al.: Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 27 (2): 295-304, 2013.
- Feng J, Tang Y: Prognostic significance of IKZF1 alteration status in pediatric B-lineage acute lymphoblastic leukemia: a meta-analysis. Leuk Lymphoma 54 (4): 889-91, 2013.
- Dörge P, Meissner B, Zimmermann M, et al.: IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 98 (3): 428-32, 2013.
- Olsson L, Castor A, Behrendtz M, et al.: Deletions of IKZF1 and SPRED1 are associated with poor prognosis in a population-based series of pediatric B-cell precursor acute lymphoblastic leukemia diagnosed between 1992 and 2011. Leukemia 28 (2): 302-10, 2014.
- Boer JM, van der Veer A, Rizopoulos D, et al.: Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia 30 (1): 32-8, 2016.
- Tran TH, Harris MH, Nguyen JV, et al.: Prognostic impact of kinase-activating fusions and IKZF1 deletions in pediatric high-risk B-lineage acute lymphoblastic leukemia. Blood Adv 2 (5): 529-533, 2018.
- Vrooman LM, Blonquist TM, Harris MH, et al.: Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv 2 (12): 1449-1458, 2018.
- Öfverholm I, Rezayee F, Heyman M, et al.: The prognostic impact of IKZF1 deletions and UKALL genetic classifiers in paediatric B-cell precursor acute lymphoblastic leukaemia treated according to NOPHO 2008 protocols. Br J Haematol 202 (2): 384-392, 2023.
- van der Veer A, Zaliova M, Mottadelli F, et al.: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 123 (11): 1691-8, 2014.
- Stanulla M, Dagdan E, Zaliova M, et al.: IKZF1plus Defines a New Minimal Residual Disease-Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J Clin Oncol 36 (12): 1240-1249, 2018.
- Mangum DS, Meyer JA, Mason CC, et al.: Association of Combined Focal 22q11.22 Deletion and IKZF1 Alterations With Outcomes in Childhood Acute Lymphoblastic Leukemia. JAMA Oncol 7 (10): 1521-1528, 2021.
- Yeoh AEJ, Lu Y, Chin WHN, et al.: Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J Clin Oncol 36 (26): 2726-2735, 2018.
- Pieters R, de Groot-Kruseman H, Fiocco M, et al.: Improved Outcome for ALL by Prolonging Therapy for IKZF1 Deletion and Decreasing Therapy for Other Risk Groups. J Clin Oncol 41 (25): 4130-4142, 2023.
- Herbrueggen H, Mueller S, Rohde J, et al.: Treatment and outcome of IG-MYC+ neoplasms with precursor B-cell phenotype in childhood and adolescence. Leukemia 34 (3): 942-946, 2020.
- Sakaguchi K, Imamura T, Ishimaru S, et al.: Nationwide study of pediatric B-cell precursor acute lymphoblastic leukemia with chromosome 8q24/MYC rearrangement in Japan. Pediatr Blood Cancer 67 (7): e28341, 2020.
- Bomken S, Enshaei A, Schwalbe EC, et al.: Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica 108 (3): 717-731, 2023.
- Liu Y, Easton J, Shao Y, et al.: The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49 (8): 1211-1218, 2017.
- Armstrong SA, Look AT: Molecular genetics of acute lymphoblastic leukemia. J Clin Oncol 23 (26): 6306-15, 2005.
- Karrman K, Forestier E, Heyman M, et al.: Clinical and cytogenetic features of a population-based consecutive series of 285 pediatric T-cell acute lymphoblastic leukemias: rare T-cell receptor gene rearrangements are associated with poor outcome. Genes Chromosomes Cancer 48 (9): 795-805, 2009.
- Mansour MR, Abraham BJ, Anders L, et al.: Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346 (6215): 1373-7, 2014.
- Steimlé T, Dourthe ME, Alcantara M, et al.: Clinico-biological features of T-cell acute lymphoblastic leukemia with fusion proteins. Blood Cancer J 12 (1): 14, 2022.
- Weng AP, Ferrando AA, Lee W, et al.: Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306 (5694): 269-71, 2004.
- Gallo Llorente L, Luther H, Schneppenheim R, et al.: Identification of novel NOTCH1 mutations: increasing our knowledge of the NOTCH signaling pathway. Pediatr Blood Cancer 61 (5): 788-96, 2014.
- Burns MA, Place AE, Stevenson KE, et al.: Identification of prognostic factors in childhood T-cell acute lymphoblastic leukemia: Results from DFCI ALL Consortium Protocols 05-001 and 11-001. Pediatr Blood Cancer 68 (1): e28719, 2021.
- Petit A, Trinquand A, Chevret S, et al.: Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 131 (3): 289-300, 2018.
- Trinquand A, Tanguy-Schmidt A, Ben Abdelali R, et al.: Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J Clin Oncol 31 (34): 4333-42, 2013.
- Bergeron J, Clappier E, Radford I, et al.: Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood 110 (7): 2324-30, 2007.
- van Grotel M, Meijerink JP, Beverloo HB, et al.: The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica 91 (9): 1212-21, 2006.
- Cavé H, Suciu S, Preudhomme C, et al.: Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: results of EORTC studies 58881 and 58951. Blood 103 (2): 442-50, 2004.
- Baak U, Gökbuget N, Orawa H, et al.: Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia 22 (6): 1154-60, 2008.
- Ferrando AA, Neuberg DS, Dodge RK, et al.: Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet 363 (9408): 535-6, 2004.
- Burmeister T, Gökbuget N, Reinhardt R, et al.: NUP214-ABL1 in adult T-ALL: the GMALL study group experience. Blood 108 (10): 3556-9, 2006.
- Graux C, Stevens-Kroef M, Lafage M, et al.: Heterogeneous patterns of amplification of the NUP214-ABL1 fusion gene in T-cell acute lymphoblastic leukemia. Leukemia 23 (1): 125-33, 2009.
- Hagemeijer A, Graux C: ABL1 rearrangements in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 49 (4): 299-308, 2010.
- Quintás-Cardama A, Tong W, Manshouri T, et al.: Activity of tyrosine kinase inhibitors against human NUP214-ABL1-positive T cell malignancies. Leukemia 22 (6): 1117-24, 2008.
- Clarke S, O'Reilly J, Romeo G, et al.: NUP214-ABL1 positive T-cell acute lymphoblastic leukemia patient shows an initial favorable response to imatinib therapy post relapse. Leuk Res 35 (7): e131-3, 2011.
- Deenik W, Beverloo HB, van der Poel-van de Luytgaarde SC, et al.: Rapid complete cytogenetic remission after upfront dasatinib monotherapy in a patient with a NUP214-ABL1-positive T-cell acute lymphoblastic leukemia. Leukemia 23 (3): 627-9, 2009.
- Crombet O, Lastrapes K, Zieske A, et al.: Complete morphologic and molecular remission after introduction of dasatinib in the treatment of a pediatric patient with t-cell acute lymphoblastic leukemia and ABL1 amplification. Pediatr Blood Cancer 59 (2): 333-4, 2012.
- Seki M, Kimura S, Isobe T, et al.: Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet 49 (8): 1274-1281, 2017.
- Nagel S, Scherr M, Kel A, et al.: Activation of TLX3 and NKX2-5 in t(5;14)(q35;q32) T-cell acute lymphoblastic leukemia by remote 3'-BCL11B enhancers and coregulation by PU.1 and HMGA1. Cancer Res 67 (4): 1461-71, 2007.
- Gutierrez A, Kentsis A, Sanda T, et al.: The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 118 (15): 4169-73, 2011.
- Ceppi F, Gotti G, Möricke A, et al.: Near-tetraploid T-cell acute lymphoblastic leukaemia in childhood: Results of the AIEOP-BFM ALL studies. Eur J Cancer 175: 120-124, 2022.
- Zhang J, Ding L, Holmfeldt L, et al.: The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481 (7380): 157-63, 2012.
- Gutierrez A, Dahlberg SE, Neuberg DS, et al.: Absence of biallelic TCRgamma deletion predicts early treatment failure in pediatric T-cell acute lymphoblastic leukemia. J Clin Oncol 28 (24): 3816-23, 2010.
- Yang YL, Hsiao CC, Chen HY, et al.: Absence of biallelic TCRγ deletion predicts induction failure and poorer outcomes in childhood T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 58 (6): 846-51, 2012.
- Montefiori LE, Bendig S, Gu Z, et al.: Enhancer Hijacking Drives Oncogenic BCL11B Expression in Lineage-Ambiguous Stem Cell Leukemia. Cancer Discov 11 (11): 2846-2867, 2021.
- Di Giacomo D, La Starza R, Gorello P, et al.: 14q32 rearrangements deregulating BCL11B mark a distinct subgroup of T-lymphoid and myeloid immature acute leukemia. Blood 138 (9): 773-784, 2021.
- Béné MC: Biphenotypic, bilineal, ambiguous or mixed lineage: strange leukemias! Haematologica 94 (7): 891-3, 2009.
- Borowitz MJ, Béné MC, Harris NL, et al.: Acute leukaemias of ambiguous lineage. In: Swerdlow SH, Campo E, Harris NL, et al., eds.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th rev. ed. International Agency for Research on Cancer, 2017, pp 179-87.
- Davies SM, Bhatia S, Ross JA, et al.: Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100 (1): 67-71, 2002.
- Krajinovic M, Costea I, Chiasson S: Polymorphism of the thymidylate synthase gene and outcome of acute lymphoblastic leukaemia. Lancet 359 (9311): 1033-4, 2002.
- Krajinovic M, Lemieux-Blanchard E, Chiasson S, et al.: Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharmacogenomics J 4 (1): 66-72, 2004.
- Schmiegelow K, Forestier E, Kristinsson J, et al.: Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: results from the NOPHO ALL-92 study. Leukemia 23 (3): 557-64, 2009.
- Relling MV, Hancock ML, Boyett JM, et al.: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93 (9): 2817-23, 1999.
- Stanulla M, Schaeffeler E, Flohr T, et al.: Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA 293 (12): 1485-9, 2005.
- Yang JJ, Landier W, Yang W, et al.: Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol 33 (11): 1235-42, 2015.
- Relling MV, Hancock ML, Rivera GK, et al.: Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst 91 (23): 2001-8, 1999.
- Moriyama T, Nishii R, Perez-Andreu V, et al.: NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet 48 (4): 367-73, 2016.
- Tanaka Y, Kato M, Hasegawa D, et al.: Susceptibility to 6-MP toxicity conferred by a NUDT15 variant in Japanese children with acute lymphoblastic leukaemia. Br J Haematol 171 (1): 109-15, 2015.
- Diouf B, Crews KR, Lew G, et al.: Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 313 (8): 815-23, 2015.
- Yang JJ, Cheng C, Yang W, et al.: Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301 (4): 393-403, 2009.
- Gregers J, Christensen IJ, Dalhoff K, et al.: The association of reduced folate carrier 80G>A polymorphism to outcome in childhood acute lymphoblastic leukemia interacts with chromosome 21 copy number. Blood 115 (23): 4671-7, 2010.
- Radtke S, Zolk O, Renner B, et al.: Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 121 (26): 5145-53, 2013.
This information does not replace the advice of a doctor. Ignite Healthwise, LLC, disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.
Page Footer
I want to...
Audiences
Secure Member Sites
The Cigna Group Information
Disclaimer
Individual and family medical and dental insurance plans are insured by Cigna Health and Life Insurance Company (CHLIC), Cigna HealthCare of Arizona, Inc., Cigna HealthCare of Illinois, Inc., Cigna HealthCare of Georgia, Inc., Cigna HealthCare of North Carolina, Inc., Cigna HealthCare of South Carolina, Inc., and Cigna HealthCare of Texas, Inc. Group health insurance and health benefit plans are insured or administered by CHLIC, Connecticut General Life Insurance Company (CGLIC), or their affiliates (see
All insurance policies and group benefit plans contain exclusions and limitations. For availability, costs and complete details of coverage, contact a licensed agent or Cigna sales representative. This website is not intended for residents of New Mexico.